Moyamoya disease
V. Přibáň1, J. Dostál1, J. Mraček1, J. Baxa2, P. Duras2
1 Neurochirurgická klinika LF UK a FN Plzeň
2 Klinika zobrazovacích metod LF UK a FN Plzeň
Cesk Slov Neurol N 2021; 84/117(2): 116–125. doi: 10.48095/cccsnn2021116
Souhrn
Nemoc moymoya je bilaterální progredující stenookluzivní postižení distální vnitřní karotidy doprovázené tvorbou bazálních kolaterál a finálně exkluzivní kolateralizací z povodí zevní karotidy. Progresi choroby postihuje angiografická klasifikace Suzukiho. Etiologie není známa, pravděpodobně se jedná o kombinaci vrozených a autoimunitních faktorů. Postihuje především asijskou populaci. Příznaky u dětských pacientů jsou ischemické, u dospělých je častým projevem krvácení. Prognóza bývá špatná. Terapie je výlučně chirurgická, buď v podobě direktního extra-intrakraniálního bypassu nebo indirektní revaskularizace, která využívá potenciál neoangiogeneze z vaskularizovaných tkání v okolí mozku. Kombinace direktní a indirektní revaskularizace představuje optimální léčbu u symptomatických pacientů.
Abstract
Moyamoya disease is bilateral progressive steno-occlusive impairment of the distal internal carotid artery accompanied by the formation of basal collaterals and finally by the exclusive collateralization from the territory of the external carotid artery. Suzuki angiographic classification describes progression of moyamoya disease. Aetiology is not known, but it is probably a combination of inherited and autoimmune factors. Asian population is mostly affected. Ischemic symptoms are typical in a pediatric population, and in adults, haemorrhage is a frequent symptom. Prognosis is poor. Therapy is exclusively surgical, either direct extra-intracranial bypass or indirect revascularization. Indirect techniques utilize potential of neoangiogenesis of vascularized tissue in the proximity of the brain. Combination of direct and indirect revascularization represents optimal treatment of symptomatic patients.
Definice
Nemoc moyamoya (moyamoya disease; MMD) je chronické progresivní stenookluzivní onemocnění terminálních úseků vnitřních karotid (internal carotid artery; ICA) a iniciálních úseků předních (anterior cerebral artery; ACA) a středních mozkových tepen (middle cerebral artery; MCA) doprovázené tvorbou bazálních kolaterál v oblasti perforátorů Willisova okruhu. Vzhled těchto kolaterál dal nemoci název. Na DSA totiž připomínají prchavý obláček cigaretového kouře – japonsky moyamoya [1]. Další kolaterály mohou být leptomeningeální a dále transdurální ze zevního karotického povodí (rete mirabile – vault moyamoya, ethmoidální moyamoya). Podmínkou definice MMD je oboustranné postižení karotického povodí a absence přidružené choroby („definitive moyamoya“). Quasi-moyamoya nebo také moyamoya syndrom je odlišná entita, u které může být jednostranné nebo oboustranné postižení v kombinaci s přidruženou chorobou. Patří sem ateroskleróza, autoimunitní onemocnění, neurofibromatóza 1. typu, Downův syndrom, primordiální nanizmus, poiradiační stav, hyperthyreóza, meningitis. Suspektní moyamoya („probable moyamoya“) znamená jednostranné postižení ICA bez přítomnosti přidružené choroby [2].
Historie
První zmínka o MMD pochází z roku 1955, kdy Shimizu a Takeuchi prezentovali na 14. sjezdu Japonské neurochirurgické společnosti ve Wakayamě angiogram bilaterální hypoplazie ICA. Následně autoři případ publikovali v roce 1957 jako kazuistické sdělení [3]. Kudo vyslovil hypotézu, že se jedná o kongenitální onemocnění. Naproti tomu Suzuki, který prezentoval na 22. kongresu Japonské neurochirurgické společnosti v roce 1963 soubor šesti pacientů, zastával teorii získaného autoimunitního onemocnění. Spor o etiologii choroby není vyřešen do současnosti. Stejný autor publikoval spolu s Takaku v roce 1969 v Archives of Neurology článek, kde v podtitulku uvedl pojem „moyamoya disease“. Editora pojem zaujal, změnil podtitulek na hlavní název a japonský výraz moyamoya se v lékařské terminologii od té doby ujal [1]. Celé jedno desetiletí panoval názor, že se jedná o chorobu čistě japonské populace. Tento názor byl vyvrácen ve druhé polovině 60. let v několika kazuistických sděleních [4–7]. Nicméně nadále platí, že incidence a prevalence v neasijské populaci je řádově nižší. Důležitým milníkem v historii MMD bylo vydání Suzukiho monografie v roce 1986. Autor v ní shrnul zkušenosti se souborem 100 pacientů a především definoval klasifikaci onemocnění dle DSA, která se užívá dodnes [8]. Detailně se věnoval experimentálnímu výzkumu nemoci a na základě výsledků tvrdil, že se jedná o autoimunitní onemocnění s iniciálním zánětem v oblasti krku a postižením krčního sympatiku autoprotilátkami. Výsledkem měly být vazospasmus a rozvoj stenookluzivního postižení terminálních ICA [9]. V terapii proto prosazoval perivaskulární sympatektomii karotidy a exstirpaci ganglion cervicalis sup. Etiologie MMD není dodnes rozluštěna, ganglionektomie a sympatektomie byly jako nástroje chirurgické léčby opuštěny.
V dalším období se rozvíjely tři strategie chirurgické léčby MMD – direktní, indirektní a kombinovaná revaskularizace. První publikace o direktní revaskularizaci technikou end-to-side bypassu mezi arteria temporalis superficialis (superficial temporal artery; STA) a kortikální větví MCA pochází od japonských autorů z roku 1978 [10]. Principem indirektní revaskularizace je vytvoření kontaktu mezi vaskularizovanou tkání a povrchem mozku s indukcí neoangiogeneze. První využil v roce 1977 Karasawa temporální sval pro encefalo-myo-synangiózu (EMS) [11]. V roce 1980 stejný autor využil k revaskularizaci omenta. Encefalo-duro-arterio-synangióza (EDAS) byla poprvé u dětí popsána v roce 1981 [12] a encefalo-duro-arterio-myo-synangióza (EDAMS) pak v roce 1993 [13].
Česká historie
Domácí literatura na téma nemoci MMD je vcelku chudá. Dosud obsahuje pouhých pět článků, z nichž poslední byl publikován v roce 2008, tedy před 12 lety. Jedná se o čtyři kazuistická sdělení a jeden soubor pacientů. Na druhou stranu je první česká práce již z roku 1970, tedy pouhý rok po publikaci MMD u pacienta mimo japonskou populaci [7]. V kazuistickém sdělení popsal Urbánek případ 7letého chlapce s klinickými projevy paroxysmů, hemiparézy a mentální retardace. V názvu práce použil dnes již zapomenuté označení „Nishimoto-Takeushi-Kudo disease“. Chlapec měl rozvinutou formu choroby s uzávěry obou ICA, bazálními kolaterálami a extrakraniálními anastomózami typu rete mirabile [14]. V neurochirurgické obci první popsal dva případy onemocnění Z. Mraček v roce 1974. Zde již byl použit termín moyamoya syndrom. V prvním případě šlo o roční dítě s pravostrannou hemiparézou, která se rozvinula po akutní nasofaryngitidě. Angiografie prokázala „triádu“ těžké stenózy obou ICA v sifonu, abnormální síť bazálních kolaterál a extracerebrálních kolaterál parietookcipitálně. Při dalším sledování dítěte byla v popředí kliniky psychomotorická retardace. Druhým pacientem byl 39letý muž s pravostrannou hemiparézou a psychickým zpomalením, který byl iniciálně hospitalizován po kraniotraumatu. Na diagnostické angiografii byl překvapivý nález bilaterální stenózy ICA a bazální kolaterály. Bohatá diskuze v článku se zabývá velmi podrobně etiologií onemocnění. Staví proti sobě kongenitální a získaný původ nemoci v kontextu dobové literatury japonských autorů. Paradoxem je, že tato problematika není dosud rozřešena. V terapii je zmíněna Suzukim propagovaná a dnes zapomenutá perivaskulární sympatektomie [15].
Sdělení Beneše staršího je z dnešního pohledu popisem quasi-moyamoya. Šestnáctiletý chlapec prodělal úraz mozku a v odstupu 5 týdnů byl hospitalizován s nálezem hemoragie v bazálních gangliích vpravo. Na DSA byl patrný spazmus pravé ICA. Dva měsíce po příhodě byla provedena kontrolní DSA s nálezem okluze ACA a MCA vpravo s přítomnými bazálními kolaterálami a rete mirabile vpravo. Klinicky byla přítomna reziduální lehká levostranná hemiparéza [16]. Kucharík et al popsali případ 3leté pacientky, u které byl klinický obraz spastické kvadruparézy, pseudobulbárního syndromu v korelaci s DSA zhodnocen jako infiltrativní tumor mozku levé hemisféry. Dívka byla ozářena a léčena cytostatiky. Diagnóza MMD byla stanovena v odstupu 8 let. Po 40 letech od prvních příznaků měla pacientka těžký mentální deficit, pseudobulbární a kvadrupyramidovou symptomatologii. Perfuze mozku byla zajištěna pouze ze zevního karotického povodí – stadium 6 dle Suzukiho [17]. Jediný článek, který zpracoval soubor pacientů, byl z pera Häckela a Beneše ml. Všichni pacienti byli dospělí. Je překvapující, že pouze v jednom případě šlo o hemoragickou formu choroby. Jestliže předchozí publikace byly deskripcí nemoci, v tomto případě pacienti absolvovali definovanou chirurgickou léčbu. Ve čtyřech případech byla provedena nepřímá revaskularizace EDAMS, v jednom případě pak kombinovaná přímá a nepřímá revaskularizace: extra-intrakraniální bypass mezi STA a kortikální větví MCA (STA-MCA bypass) + EMS. Výsledky byly popsány jako zlepšení ve čtyřech případech, stabilní stav ve čtyřech případech a úmrtí v jednom případu. Škála hodnocení uvedena nebyla. Grafické zhodnocení míry revaskularizace dokumentováno nebylo. V diskuzi byla velmi pečlivě zpracována etiologie, patologie, diagnostika a terapie na základě tehdejších znalostí [18].
Epidemiologie
Po počátečních kazuistických sděleních z 50. let a prvních větších sestavách z 60.–70. let byla určena incidence MMD v Japonsku Yamaguchim et al v roce 1980 na 0,1/100 000 [19]; o necelé desetiletí později byla stanovena incidence na 0,35/100 000 a prevalence pak na 3/100 000 [20]. Se zlepšením diagnostiky se čísla v roce 2003 navýšila na 0,54/100 000, respektive 6/100 000 [21]. Šetření z roku 2006 v populaci na ostrově Hokkaido pak prokázalo incidenci 0,94/100 000 a prevalenci 10,4/100 000. Poměr mužů a žen je ve všech sestavách přibližně 1: 2. Výskyt u bělochů je 1: 10 ve srovnání s japonskou populací. Familiární výskyt je popisován v 6–15 % [22].
Etiologie a patogeneze
Etiologie a mechanizmy patogeneze jsou nejasné. Dva základní zkoumané mechanizmy jsou genetické a zánětlivé faktory. Zastánci genetického původu nemoci uvádějí jako hlavní argumenty vysoký výskyt familiární, rasovou predilekci, oboustranné postižení a vazbu lokalizace postižení na embryonální vývoj mozkové cirkulace. Pro genetický původ svědčí objev genu RNF213 na chromozomu 17q25-ter. Polymorfizmus c.14576G>A byl popsán u 95 % pacientů s familiární formou nemoci a u 79 % pacientů se sporadickým onemocněním. Mechanizmus účinku genu není jasný, nicméně jeho výskyt koreluje s časným začátkem a těžkým průběhem choroby [23]. Vzácné čtyři polymorfizmy genu RNF213, odlišné od nálezů u asijské populace, byly nalezeny u 22 % (4 z 18 probandů) zkoumaných českých a slovenských pacientů s MMD [24].
Dále byl hodnocen vliv mutace RNF213 na RNA profil. Byla zkoumána cirkulující mikroRNA (miRNA) a ukázalo se, že miRNA deponovaná v extracelulárních vezikulách by mohla fungovat jako neinvazivní biomarker progrese MMD [25].
Velkým obhájcem teorie získaného původu MMD na podkladě autoimunitní reakce byl již Suzuki. Většina pacientů v jeho souboru měla v anamnéze zánět v oblasti orofaciální.
Zánět dle jeho teorie indukuje tvorbu humorálních protilátek, které atakují krční sympatikus. Výsledkem je pak vazospasmus a stenookluzivní proces. Postižené tepny jsou charakterizovány excentrickou hyperplazií intimy, ztenčením a nařasením lamina elastica interna a proliferací svalových buněk tunica media. Při imunohistochemickém vyšetření lze ve stěně prokázat protilátky IgG a IgM. V experimentu na japonských králících byl indukován patologický proces aplikací heterogenního séra do oblasti horního cervikálního ganglia. Protilátky IgG se prokázaly v odstupu 2 týdnů. Změny na tepnách nastaly do 12 měsíců a byly identické jako u MMD. Nález byl hodnocen jako autoimunitní vaskulitis [26].
Diagnostika a klasifikace
Základní diagnostickou modalitou je historicky angiografie, která má spolu s MR nadále ústřední postavení.
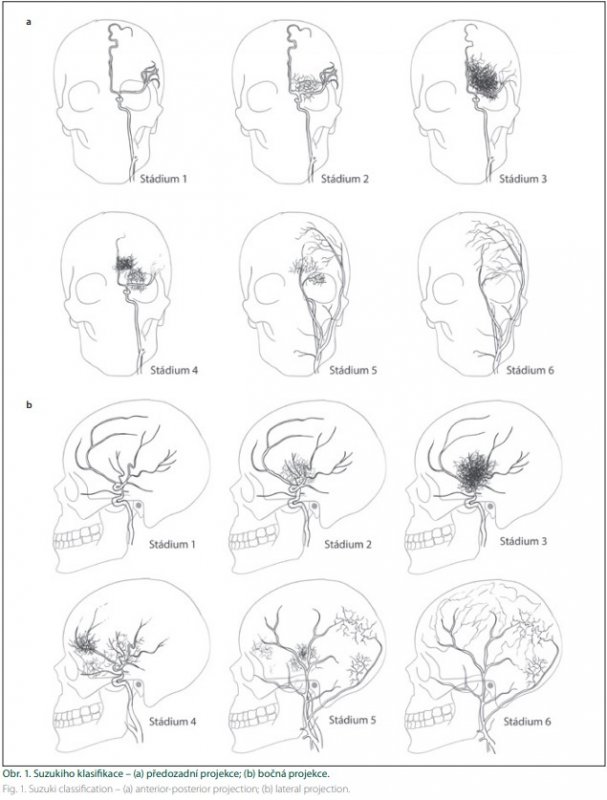
Suzuki klasifikoval ischemickou formu MMD do šesti stadií (obr. 1):
1. stadium – stenózy distální části ICA;
2. stadium – objevují se bazální kolaterály;
3. stadium – okluze MCA a ACA s bohatými bazálními kolaterálami;
4. stadium – redukce bazálních kolaterál s rozvojem tzv. ethmoidální moymoya (viz dále) a kolaterál ze zevního karotického povodí (rete mirabile);
5. stadium – postupné vymizení ICA, další redukce bazálních kolaterál a nárůst kolaterál ze zevního karotického povodí;
6. stadium – mozek je zásoben pouze cestou zevního karotického a vertebrálního tepenného povodí.
Ethmoidální moyamoya tvoří dilatované tepny, které přes strop ethmoidů přivádějí krev pro strádající mozek. Podílejí se na nich tepny jak ze zevního (a. sphenopalatina, a. facialis…), tak z vnitřního povodí karotidy (a. opthalmica). Její význam v kolateralizaci přichází ve vyšších stadiích MVD.
Při vault moyamoya (transdurální anastomózy, rete mirabile) se kolaterály se tvoří v pozdních stadiích nemoci a v posledním stadiu jsou jediným zdrojem prokrvení mozku. Suzuki popsal devět regionů transdurálních kolaterál. Pro téměř všechny oblasti je dominantním feederem a. meningea media [8].
Suzukiho klasifikace není jednoznačným popisem progrese MMD v korelaci s narůstajícími klinickými symptomy. Spíše charakterizuje kompenzační reorganizaci hemodynamiky nemoci ve smyslu konverze prokrvení cestou původně ICA ve prospěch zevního karotického povodí [27].
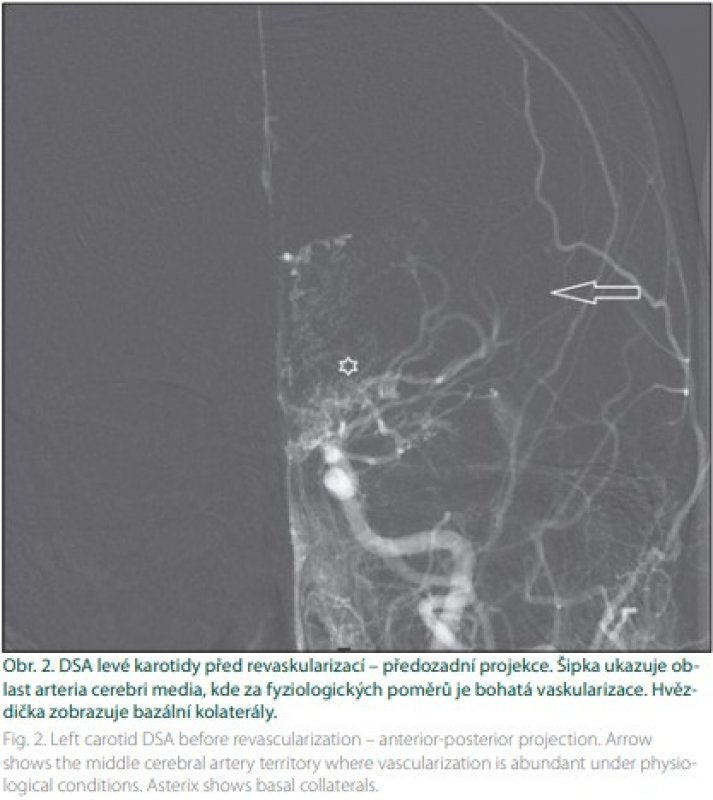
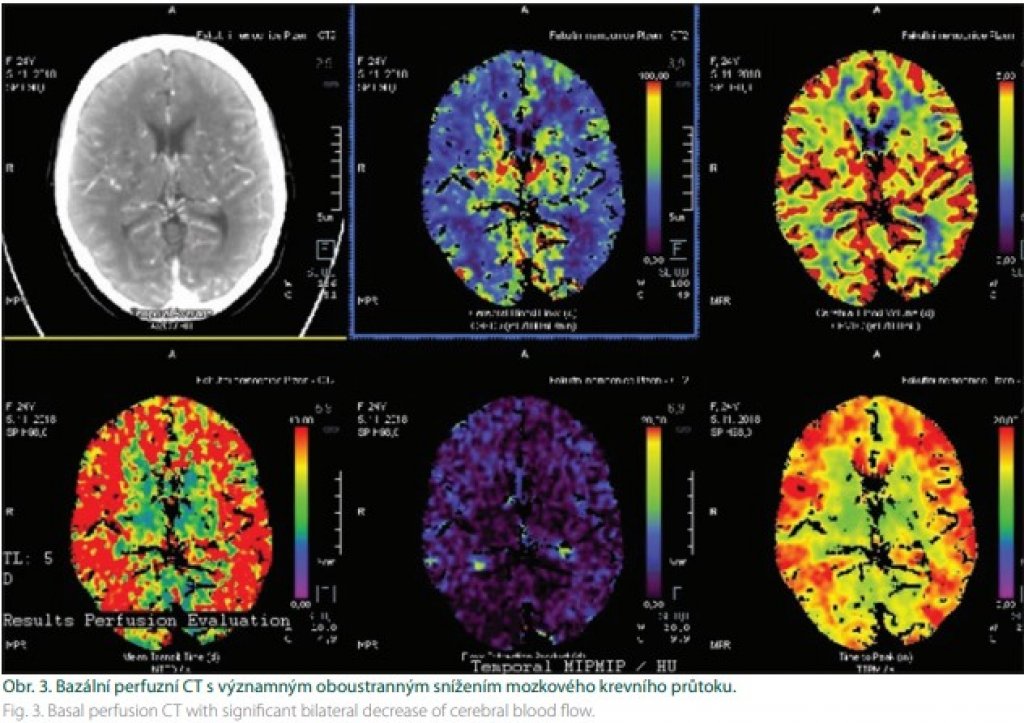
Prognóza rizika klinických projevů MMD je pochopitelně důležitá. Proto byla vytvořena Berlínská klasifikace (tab. 1), která hodnotí DSA, nález na MR a stav cerebrovaskulární rezervní kapacity (cerebrovascular reserve capacity; CVRC). Intrakraniální kompenzace na DSA znamená přítomnost leptomeningeálních a/nebo perikalózních kolaterál (obr. 2). Extrakraniální kompenzaci pak zajišťuje rete mirabile. Vyčerpání CVRC bylo v původní metodice hodnoceno na xenonCT. Nyní se provádí standardně perfuzní CT bazální a po zátěži acetazolamidem (obr. 3). Steal fenomén je pozitivní v situaci, kdy po zátěži dojde kvantitativně k poklesu průtoku o ≥ 5 %.
Na základě součtu bodů byla stadia rozdělena na lehké (1–2 body), střední (3–4 body) a těžké (5–6 bodů). Každá hemisféra byla hodnocena zvlášť. Predikce kliniky na základě Berlínské škály MMD se jeví jako přesvědčivá. Autoři měli ve svém souboru ve stadiu I 20 %, ve stadiu II 63 % a ve stadiu III 94 % symptomatických pacientů [28]. V další studii se pak zaměřili na rizika pooperační ischemie po oboustranné revaskularizaci. Riziko ischemických komplikací ve stadiu I bylo nulové, ve stadiu II 9 % a ve stadiu III pak 16 % [29].
Rizikovým faktorem krvácení u ischemické formy MMD je přítomnost chorioidálních kolaterál na DSA. Incidence krvácení je průměrně 2 % za rok. Pokud jsou přítomny chorioidální kolaterály, stoupá riziko na 5,8 %, v opačném případě je nulové [30].
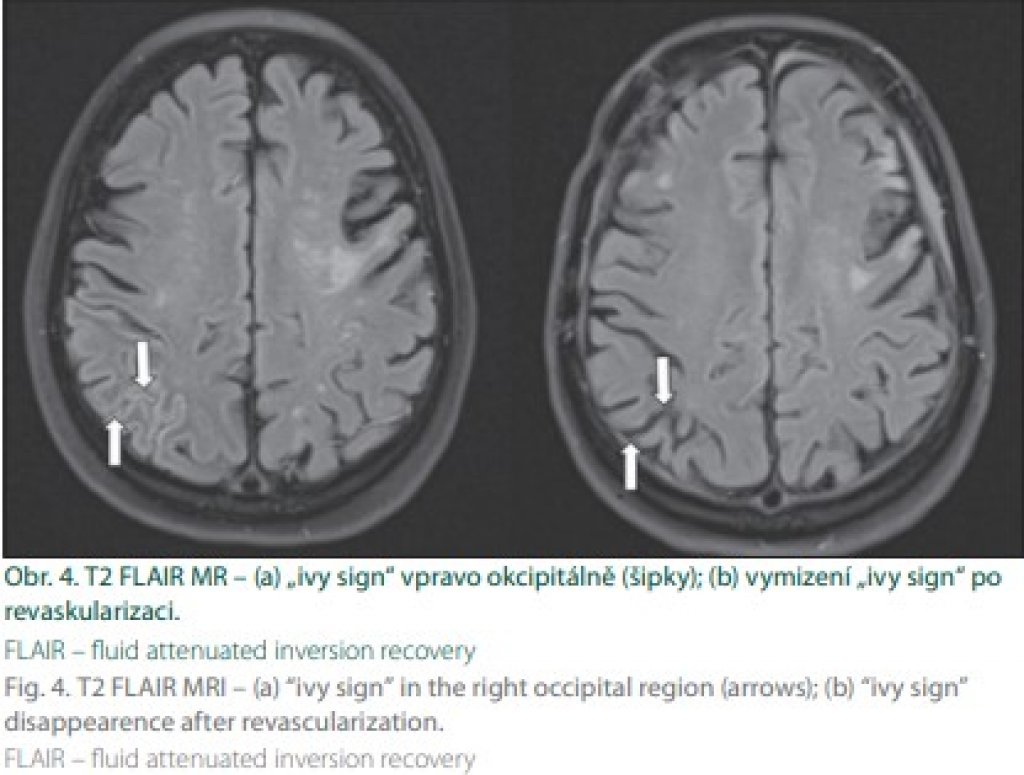
Magnetická rezonance je v současnosti klíčovou diagnostickou modalitou MMD. Difuzí vážené zobrazení prokáže akutní ischemii, obrazy T2 FLAIR (fluid attenuated inversion recovery) pak subakutní a chronické léze. V tomto vážení lze navíc prokázat tzv. „příznak břečťanu“ („ivy sign“) a „dřeňové pruhy“ („medullary streaks“). Patofyziologie jejich vzniku není zcela jasná, ale souvisí pravděpodobně s vyčerpanou CVRC. „Ivy sign“ je charakterizován pomalým až retrográdním tokem v kortikálních tepnách, „medullary streaks“ jsou lineární hyperintenzní pruhy periventrikulárně s kolmým průběhem na komoru. Po revaskularizaci dochází k vymizení těchto příznaků (obr. 4).
Susceptibilně vážené zobrazení (susceptibility weighted imaging; SWI) prokáže chronické mikrohemoragie a dilatace hlubokých medulárních periventrikulárních žil jako příznak vyčerpání CVRC. MRA v podobě 3D-TOF je neinvazivní vyšetření, které umožní hodnotit intrakraniální stenookluzivní proces ICA. 2D vyšetření s fázovým kontrastem pak navíc dokáže posoudit směr a rychlost toku krve. Hodnocení průtoku krve v mozku a posouzení CVRC umožní perfuzní vyšetření ve formě dynamického susceptibilního kontrastu (dynamic susceptibility contrast; DSC) s použitím gadolinia nebo arterial spin labeling (ASL) bez využití kontrastní látky [31].
Klinický obraz
Nemoc moyamoya má bimodální charakter věkového rozvrstvení. Maximum výskytu nemoci je ve věku 4–10 let a poté ve 40–45 letech. Pediatričtí pacienti mají typicky příznaky ischemické. Charakterizují je ischemický iktus, tranzitorní ischemické ataky (TIA) izolované nebo repetitivní, křeče, cefalea a choreatické pohyby.
U dospělých pacientů je typickým projevem krvácení. Příčinou je ruptura aneuryzmat vznikajících v důsledku hemodynamického stresu v patologicky změněných cévách u MMD. Aneuryzmata mohou vzniknout na Willisově okruhu, distálně na chorioidálních tepnách a na bazálních kolaterálách. Ruptura aneuryzmat je příčinou intracerebrálního krvácení v bazálních gangliích, talamu a komorách. I u dospělých může však být stejně jako u dětí projevem MMD ischemický iktus.
Pro asymptomatické MMD nejsou stanoveny jednoznačné doporučující postupy. Někteří autoři indikují revaskularizaci v případě pokročilých stenookluzivních nálezů, podpořených vyčerpáním cerebrovaskulární reaktivity. Jiní mají aktivní přístup a provádějí paušální revaskularizaci s argumentem špatného přirozeného průběhu nemoci [28].
Terapie
Přirozený průběh MMD je devastující. Riziko iktu je udáváno až 13 % za rok [32]. Konzervativní terapie je neefektivní [33]. Neúčinnost byla prokázána i v případě antiagregační léčby [34].
Endovaskulární léčba stenookluzivní formy angioplastikou je neúspěšná, restenóza tepny je pravidlem. V současnosti panuje konsenzus, že jedinou léčebnou modalitou je chirurgická léčba. U krvácení je indikována evakuace hematomu při hemocefalu v kombinaci s komorovou drenáží. Proximálně uložená aneuryzmata jsou vyřazena z cirkulace chirurgickou nebo intervenční technikou.
V principu lze rozdělit chirurgické techniky léčby stenookluzivní formy MMD na direktní, indirektní a kombinovanou revaskularizaci.
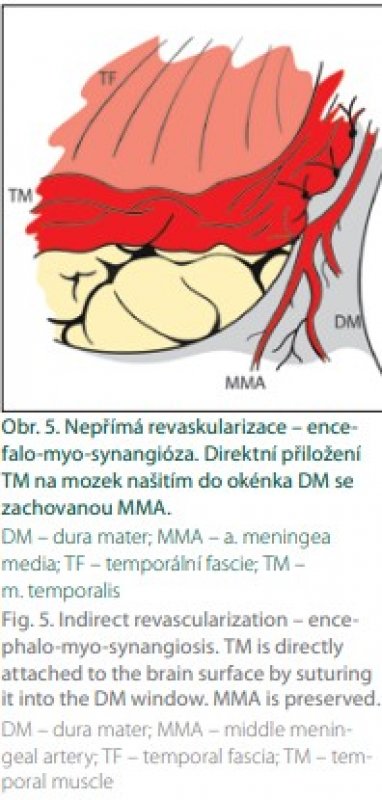
Indirektní revaskularizace znamená přiložení vaskularizované tkáně na povrch mozku s následnou neoangiogenezí a prorůstáním kapilár do kortexu. Patofyziologickým podkladem tohoto jevu je přítomnost cévních růstových faktorů u pacientů s MMD. Mezi růstové cévní faktory řadíme vaskulární endoteliální růstový faktor, matrix metaloproteinázu-9, hepatocytární růstový faktor a interleukin 1. Přítomnost faktorů je exkluzivní pro MMD, u aterosklerózy se cévní faktory nevyskytují, a proto je u ní indirektní revaskularizace neefektivní. Jako zdroj revaskularizace se využívá bohatě vaskularizovaná tkáň v dostupné blízkosti povrchu mozku: temporální sval, dura mater s a. meningea media, periost, galea, popřípadě omentum. První pokusy o nepřímou revaskularizaci pocházejí od Kredela z roku 1942. První indirektní operaci pro MMD provedl Karasawa v roce 1977. Šlo o techniku EMS. Mobilizoval temporální sval, který poté našil k okrajům „okénka“ v dura mater (obr. 5). Během několika měsíců došlo k „vrůstu“ kapilár ze svalu do kortexu s následným zlepšením krevního průtoku [11].
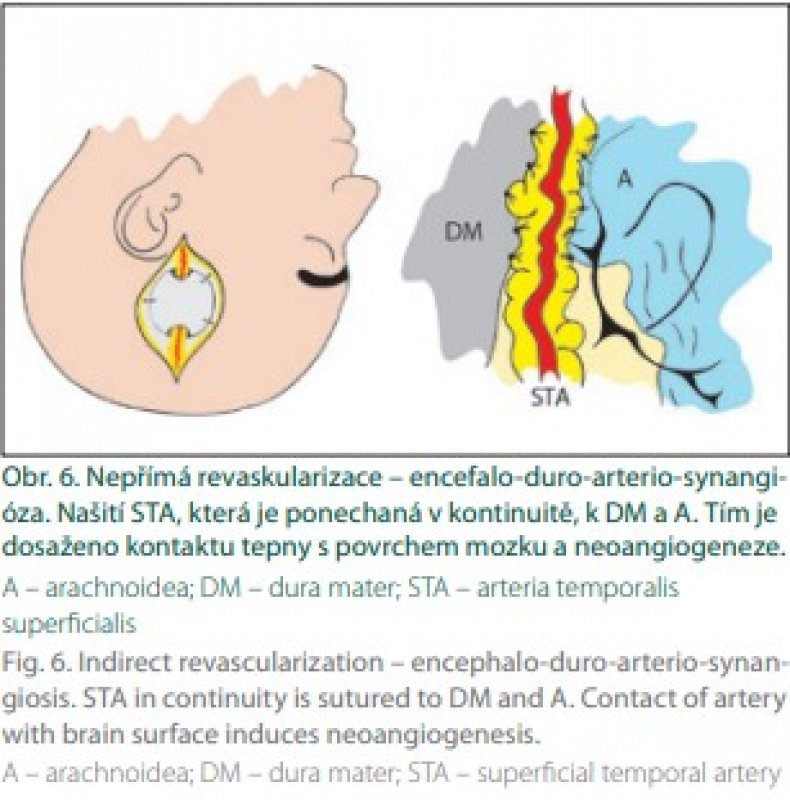
V roce 1980 stejný autor využil k revaskularizaci omenta [35]. Tato technika je dnes prakticky opuštěna. Následně byly využity další tkáně jako zdroj novotvorby kapilár EDAS (obr. 6) [12] a EDAMS pak v roce 1993 [13].
Efektivita výše popsaných technik selhává, pokud je hypoperfuze i v teritoriu přední mozkové tepny. Proto zavedl Kawaguchi v roce 1996 techniku mnohočetných návrtů. Do otvoru v dura mater a arachnoidee byl v každém návrtu vložen periost [36]. Metoda má v současnosti řadu odpůrců. Z pohledu komplexního řešení rozsáhlé hypoperfuze se jeví jako perspektivní kombinovaná technika encefalo-duro-myo-angio-periosteo-synangiózy (EDMAPS) [37]. V principu přibývá revaskularizace teritoria ACA pomocí periostu.
Obecnou nevýhodou nepřímé techniky je delší časový interval do efektivní revaskularizace. Výhodou pak je nižní technická náročnost ve srovnání s bypassem a menší riziko závažných perioperačních komplikací, zejména ischemie a hyperperfuzního syndromu. Nepřímá revaskularizace má své pevné místo zejména u dětské populace, kde může být technické provedení bypassu obtížné až nemožné. Dlouhodobé výsledky jsou přesvědčivé. Scott ve své sestavě 55 pacientů s průměrným věkem 6,2 roku registroval po 20 letech sledování iktus v 1,7 %. Celkem 86 % pacientů mělo stejné nebo zlepšené skóre modifikované Rankinovy škály. U všech provedl modifikovanou EDAS – piální synangiózu [38]. Podobně přesvědčivé výsledky prezentoval na 450 případech Steinberg. Direktní revaskularizaci provedl v 95 % u dospělých, a v 76 % u dětských pacientů. Perioperační morbidita/mortalita byla 4,2 %, kumulativní 5leté riziko iktu/smrti pak 5,5 % [39].
Direktní revaskularizace – extra-intrakraniální bypass – přináší okamžité zlepšení krevního průtoku [29]. Výsledkem je úprava cerebrovaskulární reaktivity a snížení rizika iktu. Následná redukce bazálních kolaterál je pravděpodobně příčinou významného snížení rizika krvácení u MMD, jak bylo prokázáno v multicentrické randomizované studii [40].
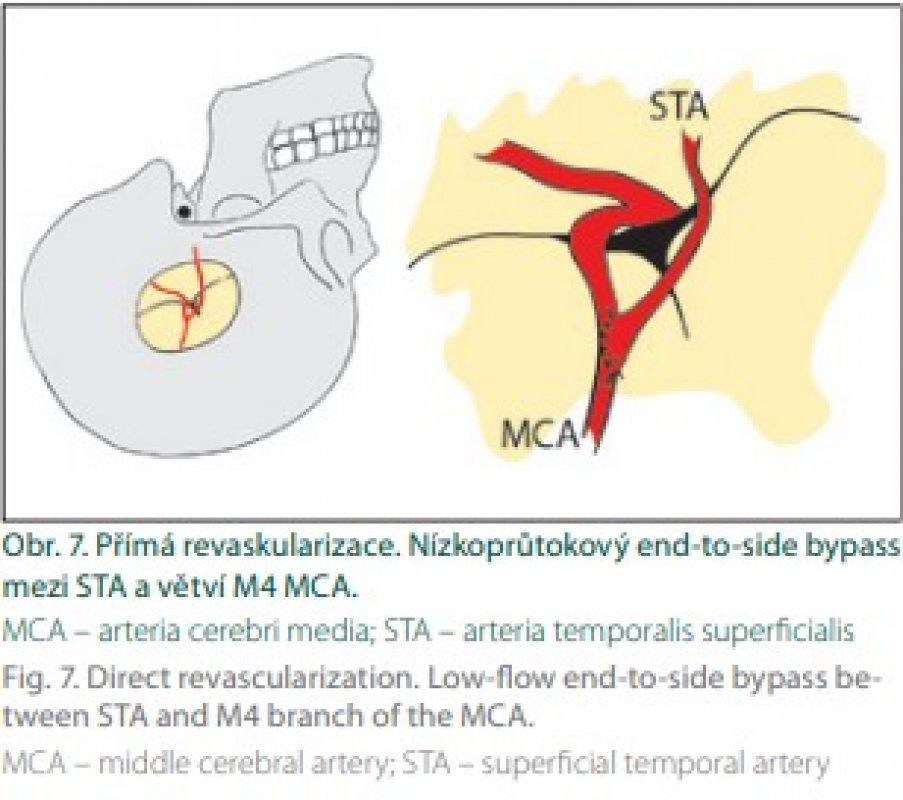
První publikace o direktní revaskularizaci technikou end-to-side bypassu mezi STA a kortikální větví MCA pochází od japonských autorů [10] (obr. 7).
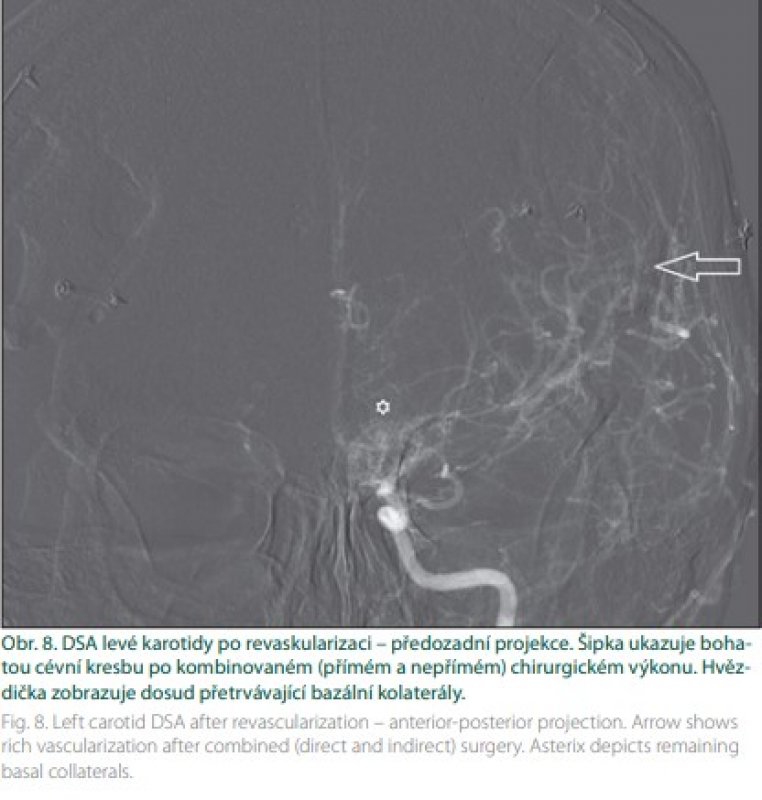
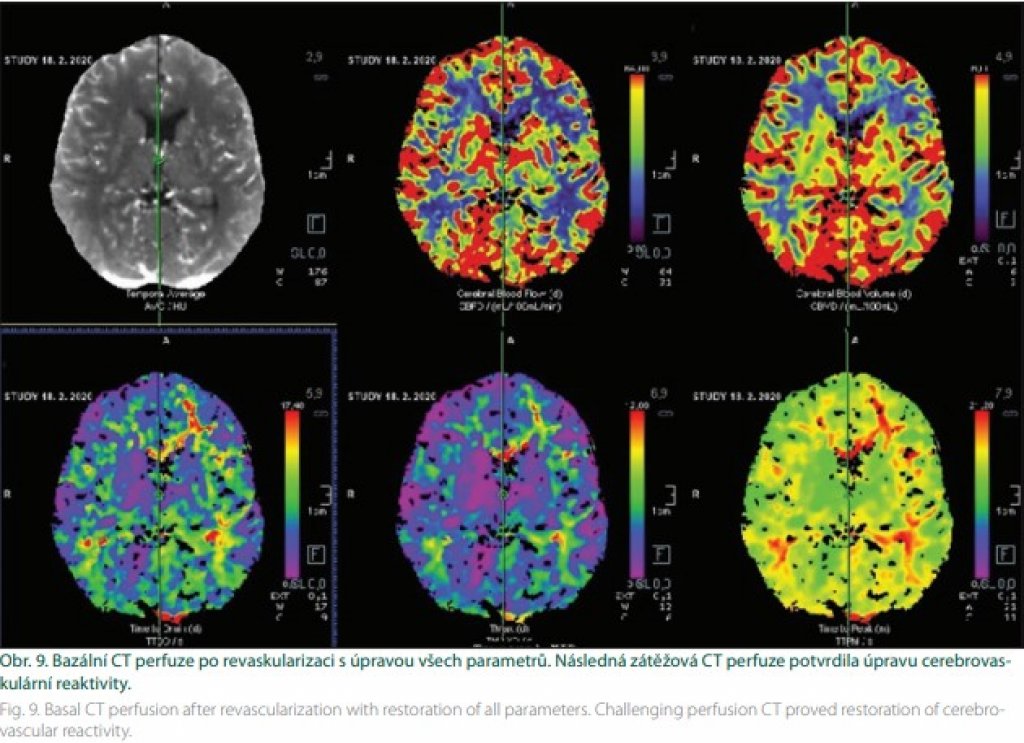
Kombinovaná revaskularizace znamená využití bypassu spolu s jednou technikou nepřímé revaskularizace (obr. 8). Vajkoczy využívá u symptomatických hemisfér kombinovanou revaskularizaci, na straně asymptomatické pak revaskularizaci nepřímou. Oba výkony provádí v jedné době [41]. Během výkonu je pacient vystaven riziku ischemie. V její prevenci je vhodné dodržovat normotenzi, respektive mírnou arteriální hypertenzi během výkonu, normokapnii a normální tělesnou teplotu. Během sutury je vhodné zvednout frakci O2 na 100 %. Náhlé zvýšení průtoku v oblastech s vyčerpanou reaktivitou přináší riziko hyperperfuzního syndromu. Velké riziko hyperperfuze je zejména u pacientů s opakovanými TIA. Diagnózu stanoví perfuzní CT (obr. 9). V terapii má ústřední místo kontrola/redukce krevního tlaku [42].
Závěr
Nemoc moyamoya je oboustranné progresivní okluzivní postižení distálních úseků ICA s novotvorbou bazálních kolaterál. Etiologie MMD je dosud nejasná. Pravděpodobně se jedná o kombinaci vrozených a imunitních faktorů. Postihuje dominantně, nikoliv výlučně, asijskou populaci. Příznaky u dětských pacientů jsou ischemické, u dospělých je častým projevem krvácení. Terapie ischemické formy je výlučně chirurgická v podobně direktní, indirektní a kombinované revaskularizace. Při krvácivých projevech je cílem evakuace hematomu, drenáž komor v případě hemocefalu a ošetření prasklých aneuryzmat.
Grantová podpora
Práce byla podpořena MZ ČR – RVO (Fakultní nemocnice Plzeň – FNPl, 00669806).
Poděkování
Poděkování Mgr. P. Řihánkovi za zpracování obrazové dokumentace.
doc. MUDr. Vladimír Přibáň, Ph.D.
Neurochirurgická klinika
LF UK a FN Plzeň
E. Beneše 1128
301 00 Plzeň
e-mail: pribanv@fnplzen.cz
Přijato k recenzi: 1. 10. 2020
Přijato do tisku: 30. 3. 2021
Literatura
1. Suzuki J, Takaku A. Cerebrovascular „moyamoya“ disease. Disease showing abnormal net-like vessels in base of brain. Arch Neurol 1969; 20(3): 288–299. doi: 10.1001/archneur.1969.00480090076012.
2. Research Committee on Pathology and Treatment of Spontaneous Occlusion of the Circle of Willis; Health Labour Sciences Research Grant for Research on Measures for Intractable Diesases: Guidelines for diagnosis and tratment of moyamoya disease (spontaneous occlusion of the circle of Willis). Neurol Med Chir (Tokyo) 2012; 52(5): 245–266. doi: 10.2176/nmc.52.245.
3. Takeuchi K, Shimizu K. Hypoplasia of the bilateral internal carotid arteries. Brain Nerve 1957; 9: 37–43.
4. Pool JL, Wood EH, Maki Y. On the cases with abnormal vascular network in the cerebral basal region in the United States. Neurol Med Chir 1966; 8: 255–258.
5. Simon J, Sabouraud O, Guy G et al. Un cas de maladie de Nishimoto. A propos d’une maladie rare et bilaterale de la carotide interne. Rev Neurol 1968; 119(4): 376–383.
6. Busch HF. Unusual collateral circulation in a child with cerebral arterial occlusion. Psychiat Neurol Neurochir 1969; 72(1): 23–28.
7. Taveras JM. Multiple progressive intracranial arterial occlusion: a syndrome of children and young adults. Am J Roentgenol Radium Ther Nucl Med 1969; 106(2): 235–268. doi: 10.2214/ajr.106.2.viii.
8. Suzuki J. Cerebral angiography. In: Suzuki J (ed). Moyamoya disease. Berlin, Heidelberg, New York, Tokio: Springer-verlag 1986: 17–52.
9. Suzuki J. Etiology. In: Suzuki J (ed). Moyamoya disease. Berlin, Heidelberg, New York, Tokio: Springer-verlag 1986: 131–143.
10. Karasawa J, Kikuchi H, Furuse S et al. Treatment of moyamoya disease with STA-MCA anastomosis. J Neurosurg 1978; 49(5): 679–688. doi: 10.3171/jns.1978.49.5.0679.
11. Karasawa J, Kikuchi H, Furuse S et al. A surgical treatment of moymamoya disease „encephalo-myo-synangiosis“. Neurol Med Chir (Tokyo) 1977; 17(1 Pt 1): 29–37. doi: 10.2176/nmc.17pt1.29.
12. Matsushima Y, Fukai N, Tanaka K et al. A new surgical treatment of moyamoya disease in children: a preliminary report. Surg Neurol 1981; 15(4): 313–320. doi: 10.1016/s0090-3019 (81) 80017-1.
13.Kinugasa K, Mandai S, Kamata I et al. Surgical treatment of moyamoya disease: operative technique for encephalo-duro-arterio-myo-synangiosis, its follow-up, clinical results, and angiograms. Neurosurgery 1993; 32(4): 527–531. doi: 10.1227/00006123-199304000-00006.
14. Urbánek K, Fárková H, Klaus E. Nishimoto-Takeuchi-Kudo disease: case report. J Neurol Neurosurg Psychiat 1970; 33(5): 671–673. doi: 10.1136/jnnp.33.5.671.
15. Mraček Z, Kohoutek V. Syndrom moyamoya. Cas Lek Cesk 1974; 113(50–51): 1561–1564.
16. Beneš V st. Syndrom moyamoya. Cesk Pediatr 1982; 37: 674.
17. Kucharík M, Roth J, Faltýnová E et al. Průběh onemocnění moyamoya u pacientky sledované od 3 let do 40 let věku. Neurol praxi 2008; 9(1): 49–51.
18. Häckel M, Beneš V ml. Onemocnění moyamoya. Přehled a soubor 9 nemocných. Cesk Slov Neurol N 1997; 60/93(2): 142–151.
19. Yamaguchi T, Tashiro M, Minematsu K et al. Summary of Japanese survey of occlusion of the circle of Willis. In Reports by the research committee on spontaneous occlusion of the circle of Willis. Tokyo: Japanese ministry of Health and welfare 1955: 13–22.
20. Ikezaki K, Inamura T, Kawano T et al. Clinical features of probable Moyamoya disease in Japan. Clin Neurol Neurosurg 1997; 99 (Suppl 2): S173–S177. doi: 10.1016/s0303-8467(97)00053-x.
21. Baba T, Houkin K, Kuroda S. Novel epidemiological features of Moyamoya disease. J Neurol Neurosurg Psychiatry 2008; 79(8): 900–904. doi: 10.1136/jnnp.2007.130666.
22. Shuo H, Zhen NG, Mingchao S et al. Etiology and pathogenesis of Moyamoya disease: an update on disease prevalence. Int J Stroke 2017; 12(3): 246–253. doi: 10.1177/1747493017694393.
23. Kamada F, Aoki Y, Narisawa A et al. A genome-wide association study identifies RNF 213 as the first Moyamoya disease gene. J Hum Genet 2011; 56(1): 34–40. doi: 10.1038/jhg.2010.132.
24. Kobayashi H, Brozman M, Kyselová K et al. RNF213 Rare variants in Slovakian and Czech moyamoya disease patients. PLoS ONE 2016; 11(10): e0164759. doi: 10.1371/journal.pone.0164759.
25. Lee MJ, Falllen S, Zhou Y et al. The impact of moyamoya disease on RNF 213 mutations on the spectrum of plasma protein and microRNA. J Clin Med 2019; 8(10): 1648–1667. doi: 10.3390/jcm8101648.
26. Zhang H, Rao M, Zhang S. An experimental study on the etiology and pathogenesis of Moyamoya disease. Chin J Neurol 1996; 29: 178–181.
27. Fujimura M, Tominaga T. Diagnosis of moyamoya disease: international standard and regional differences. Neurol Med Chir (Tokyo) 2015; 55(3): 189–193. doi: 10.2176/nmc.ra.2014-0307.
28. Czabanka M, Peňa-Tapia P, Schubert GA et al. Proposal for a new grading of moyamoya disease in adult patients. Cerebrovasc Dis 2011; 32(1): 41–50. doi: 10.1159/000326077.
29.Acker G, Fekonja L, Vajkoczy P. Surgical management of moyamoya disease. Stroke 2018; 49(2): 476–482. doi: 10.1161/STROKEAHA.117.018563.
30. Funaki T, Takahashi JC, Houkin K et al. Effect of chorioidal collaterall vessels on de novo hemorrhage in moyamoya disease: analysis of nonhemorrhagic hemisferes in the Japan Adult Moyamoya Trial. J Neurosurg 2020; 132(2): 408–414. doi: 10.3171/2018.10.JNS181139.
31. Lehman VT, Cogswell PM, Rinaldo L et al. Contemporary and emerging magnetic resonance imaging nethods for evaluation of moyamoya disease. Neurosurg Focus 2019; 47(6): E6. doi: 10.3171/2019.9.FOCUS19616.
32. Kuroda S. AMORE Study Group. Asymptomatic moyamoya disease. Literature review and ongoing AMORE study. Neurol Med Chir (Tokyo) 2015; 55(3): 194–198. doi: 10.2176/nmc.ra.2014-0305.
33. Choi JU, Kim DS, Kim EY et al. Natural history of moyamoya disease: comparison of aktivity of daily living in surgery and non surgery group. Clin Neurol Neurosurg 1997; 99 (2 suppl): S11–S18.
34. Yamada S, Oki K, Itoh I et al. Research Committee on Spontaneous Occlusion fo Circle of Willis (Moyamoya Disease). Effect of surgery and antiplatelet therapy in ten-year follow-up from registry study of research committee on moyamoya disease in Japan. J Stroke Cerebrovasc Dis 2016; 25(2): 340–349. doi: 10.1016/j.jstrokecerebrovasdis.2015.10.003.
35. Karasawa J, Kikuchi H, Kavamura J et al. Intracranial tranplantation of the omentum for cerebrovascular moyamoya disease: a two-year follow-up study. Surg Neurol 1980; 14(6): 444–449.
36. Kawaguchi T, Fujita S, Hosoda K et al. Multiple burr-hole operation for adult moyamoya disease. J Neurosurg 1996; 84(3): 468–474. doi: 10.3171/jns.1996.84.3.0468.
37. Kuroda S, Houkin K, Ishikawa T et al. Novel bypass surgery for moyamoya disease using pericranial flap: its impact on cerebral hemodynamics and long-term follow-up. Neurosurgery 2010; 66(6): 1093–1101. doi: 10.1227/01.NEU.0000369606.00861.91.
38. Riordan CP, Storey A, Cote DJ et al. Results of more than 20 years of follow-up in pediatric patients with moyamoya disease undergoing pial synangiosis. J Neurosurg Ped 2019; 23: 586–592. doi: 10.3171/2019.1.PEDS18457.
39. Guzman R, Lee M, Achrol A et al. Clinical outcome after 450 revascularization procedures for moyamoya disease. J Neurosurg 2009; 111(5): 927–935. doi: 10.3171/2009.4.JNS081649.
40. Miyamoto S, Yoshimoto T, Hashimoto N et al. Effects of extracranial-intracranial bypass for patients with hemorrhagic moyamoya disease: results of the Japan Adult Moyamoya Trial. Stroke 2014; 45(5): 1415–1421. doi: 10.1161/STROKEAHA.113.004386.
41. Acker G, Fekonja L, Vajkoczy P. Surgical management of moyamoya disease. Stroke 2018; 49(2): 476–482. doi: 10.1161/STROKEAHA.117.018563.
42. Föhre B, König S. Perioperative management and considerations. In: Vajkoczy P (ed). Surgical techniques in moyamoya vasculopathy. New York: Thieme 2020: 2–7.
Článek byl uveřejněn s laskavým svolením redakce Neurologie a neurochirurgie.
www.csnn.eu