Beta-thalassemia minor and maior in pregnancy
Racková J.¹,², Mrázek M.²
¹Gynekologicko-porodnická klinika 1. LF UK a nemocnice Na Bulovce, Praha
²Gynem s. r. o.
Transfuze Hematol Dnes 2022; 28(4): 228–232doi: 10.48095/cctahd2022228
SOUHRN: Prezentujeme kazuistiku 35leté primigravidy/nulipary, která je nosičkou recesivní formy β-talasémie. Před první návštěvou v naší ambulanci byla diagnóza známá od dětství, a to na základě vyšetření krevního obrazu a rodinné anamnézy, nicméně nebyla geneticky potvrzena. Partner měl v dětství anémii a taktéž u něj byla potvrzena heterozygotní forma β-talasémie. Oběma bylo pomocí DNA-sekvenování verifikováno nosičství β-talasémie s mutací IVS I-6. T>C až během probíhající gravidity. Prvotrimestrální screening plodu byl negativní, z biopsie choriových klků a následného genetického vyšetření se však potvrdila homozygotní varianta β-talasémie. β-thalassemia maior je závažné vrozené onemocnění s nejvyšším výskytem v subtropických geografických oblastech. I přes velký pokrok v terapii zůstává stále vysoká morbidita i mortalita postižených jedinců. Díky globálním společenským změnám a migraci se u nás mohou objevovat choroby, které se dříve ve střední Evropě nevyskytovaly. Zdravotnictví by mělo být připravené na nové výzvy a dokázat jim čelit účinnou primární i sekundární prevencí vč. prenatální diagnostiky.
SUMMARY: We present a case of a 35-year-old primigravid nulliparous woman, which is a carrier of recessive form of beta thalassemia. Prior to the first visit at our clinic, the diagnosis was not genetically confirmed, it was based only on the blood-count in the childhood. Her partner also had childhood anaemia, and heterozygous form was suspected. They both underwent a genetic counselling; both were diagnosed with beta thalassemia minor by DNA sequencing. The first trimester foetal screening was negative, but the homozygous form of beta thalassemia was confirmed from a chorionic villus biopsy and subsequent genetic examination. Beta thalassemia maior is a serious congenital autosomal recessive disease with the highest incidence in subtropical geographical areas. Despite great progress in therapy, the morbidity and mortality of affected individuals remains high. Due to global social changes and migration, genetic rare diseases that did not occur in Central Europe may emerge. Healthcare should be well prepared and be able to cope with an effective primary and secondary prevention including the prenatal testing.
ÚVOD
Hemoglobinopatie se celosvětově řadí mezi nejčastější vrozené recesivně monogenní poruchy krvetvorby, jejichž závažné formy jsou spojeny s chronickými invalidizujícími a život ohrožujícími zdravotními důsledky. V ČR bylo v roce 2017 diagnostikováno téměř 900 nemocných s různými typy hemoglobinopatií, což klade zvýšené nároky na jejich správnou diagnostiku vč. diagnostiky prenatální [1]. Syntéza hemoglobinu, probíhající během erytropoézy, pozůstává z celé řady kroků. Globin stejně jako jiné bílkoviny vzniká na ribozomech, produkce hemu probíhá v mitochondriích a v cytoplazmě. U dospělých jedinců je adultní hemoglobin HbA zastoupen v 97 % a skládá se ze dvou podjednotek alfa a dvou beta. Hemoglobin HbA2 je v dospělosti zastoupen ve 2 % a skládá se ze dvou alfa a dvou delta podjednotek. Reziduálně, a to v méně než 1 %, je zastoupen také fetální hemoglobin HbF skládající se ze dvou alfa a dvou gama globinových podjednotek. Struktura hemoglobinu patří mezi nejprostudovanější globulinové proteiny – za popsání struktury hemoglobinu dostali M. Perutz a J. Kendrew v roce 1962 Nobelovu cenu za chemii.
Mezi hemoglobinopatie patří řada vrozených onemocnění, která jsou charakteristická poruchou syntézy globinových řetězců. Dělíme je na: kvalitativní – způsobující strukturální změny hemoglobinu. Jedná se např. o srpkovitou anémii, kde defektní produkce hemoglobinu S tvoří v erytrocytech agregáty a erytrocyty mají typický srpkovitý tvar, nebo hemoglobinopatii se zvýšenou afinitou hemoglobinu ke kyslíku, způsobující erytrocytózu, či hemoglobinopatii s nestabilitou Hb, způsobující hemolýzu.
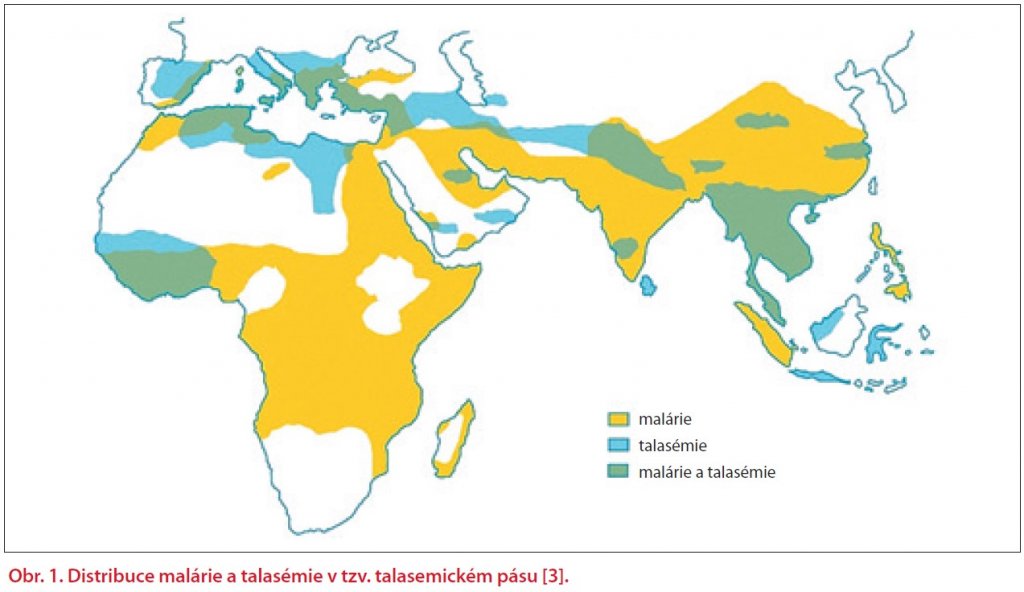
Kvantitativní hemoglobinopatie se projevují snížením nebo absencí exprese některého z globinových genů. Dle poruchy syntézy globinového řetězce je dělíme na talasémie alfa (α) a beta (β) a další talasemické syndromy. Dochází k bodovým mutacím, inzercím nebo delecím na chromozomu 16 (geny pro α-globin) nebo 11 (geny pro β-globin). Závažnost klinických projevů talasémií závisí na velikosti nerovnováhy α- a β-globinových řetězců. Předpokládá se, že se důsledkem selekčního tlaku Plasmodium falciparum vyvinula částečná protektivní ochrana proti malárii. Talasémie byla identifikována z kosterních ostatků Homo sapiens sapiens starých až 7 000 let. Původ slova thalassaemia je z 30. let, z řeckého thalassos – moře a haima – krev. Geograficky tzv. talasemický pás tvoří země středního východu, severní Afriky, jižní Ameriky, Indie a jihovýchodní Asie (především Vietnam, Čína, Taiwan, Singapur, Filipíny a Indonésie). V Evropě se mezi nejpostiženější regiony počítá Sardinie, Řecko, Turecko a Kypr, kde počet přenašečů dosahuje až 15 %. Celosvětově je přibližně 90 milionů populace (1,5 %) přenašeči talasemických alel. Každoročně se narodí okolo 60 000 symptomatických jedinců. Celková roční incidence symptomatické β-talasémie v Evropské unii je 1: 10 000 obyvatel. V rozvojových zemích se odhaduje až 100 000 případů dětských úmrtí na homozygotní formu β-talasémie ročně [2] (obr. 1) [3].
β-talasémie vznikají v důsledku poruchy tvorby β-globinových řetězců. Podle typu mutací dochází k rozsáhlé heterogenitě molekulárních defektů β-globinových řetězců, od absolutní nepřítomnosti změn v krevním obrazu až po různorodé poruchy syntézy [4]. Obecně se klasifikují do tří hlavních podskupin (β-thalassemia maior, intermedia a minor), které se liší genotypem i fenotypem a většinou se dědí podle Mendelovské genetiky.
β-thalassemia maior je homozygotní forma, která se projeví během prvního až několika dalších měsíců po narození. Je slučitelná se životem a nedá se odhalit prenatální ultrazvukovou diagnostikou (na rozdíl např. od homozygotní α-talasémie způsobující Bartsův hydrops fetalis). Je narušena struktura β-globinového řetězce hemoglobinu a dochází k sníženému množství (β-plus) nebo úplné absenci (β-nula) globinového řetězce.
Homozygotní jedinci s β-nula variantou trpí relativním nadbytkem α-globinových řetězců, které se během erytropoézy sráží v erytroidních prekurzorech v kostní dřeni. V důsledku toho dochází k jejich předčasné buněčné smrti a anémii, která dále stimuluje produkci erytropoetinu a způsobuje zvýšení počtu retikulocytů. U neléčených dětí s β-thalassemia maior dochází k neefektivní expanzi kostní dřeně a postižení jedinci trpí kostními deformitami. U dětí jsou typické Cooleyho anémie a facies thalassemica. Extramedulární hematopoéza způsobí následnou hepatosplenomegalii, a dochází tak k růstové retardaci, anoxii, bledosti, ikteru a snížení svalové hmoty. V důsledku opakovaných transfuzí a nízké hladiny hepcidinu trpí pacienti relativním nadbytkem železa způsobujícím endokrinní dysfunkce vč. pubertas tarda, diabetes mellitus, dysfunkce štítné žlázy, dilatační myokardiopatii nebo jaterní fibrózu. Mezi terapeutické možnosti patří opakované transfuze, chelatační léčba, podávání hydroxyurey (vedoucí ke zvýšení tvorby fetálního hemoglobinu), v rozvinutých zemích alogenní transplantace hematopoetických kmenových buněk nebo na experimentální úrovni genová terapie [4].
β-thalassemia intermedia je fenotypově heterogenní skupinou onemocnění. Částečná produkce β-globinových řetězců je zachována, dle postižení mohou být jedinci asymptomatičtí nebo mohou vyžadovat trvalou transfuzní léčbu. Je prezentována mikrocytární anémií, pacienti trpí extramedulární hematopoézou se splenomegalií, kostními deformitami, bolestivými ulceracemi dolních končetin a sklonem k trombóze [5].
β-thalassemia minor je převážně asymptomatická forma u heterozygotních přenašečů. V naší populaci je to nejčastější forma, typická je lehká anémie s mikrocytózou (MCV 60–70 fl), hypochromazií (MCH < 27 pg) a s erytrocytózou (> 5×1012/l) při normální hladině sérového železa. Nemocní nevyžadují transfuzní terapii. V rámci diferenciální diagnostiky mikrocytární hypochromní anémie, která je častá v těhotenství, vyloučíme nedostatek železa, při elektroforéze hemoglobinu bývá zvýšená hladina HbA2 a někdy i HbF, erytrocytóza. Neméně důležitá je také rodinná anamnéza. Pro dovyšetření lze pomocí molekulárně-genetických vyšetření určit typ mutace [1].
VLASTNÍ POZOROVÁNÍ
Pětatřicetiletá pacientka, primigravida/nulipara, původem z Portugalska dlouhodobě pracující v ČR se dostavila k registraci k preventivní gynekologické prohlídce. Při odběru anamnézy, která je pro preventivní gynekologicko-porodnickou péči klíčová, pacientka udávala β-talasémii minor v souvislosti s výskytem anémie v dětství. Nebyla testována geneticky. Oba její bratři jsou nosiči β-thalassemia minor, mají zdravé děti. Na cílený dotaz, zda partner má také β-talasémii, pacientka odpověděla kladně. Páru bylo doporučeno hematologické a genetické vyšetření a byli poučeni o možnosti preimplantační genetické diagnostiky (PGD) při technikách in vitro fertilizace (IVF). Po 3 měsících pacientka přišla po spontánní koncepci v 6. týdnu těhotenství bez komplikací, avšak žádné vyšetření k potvrzení diagnózy hemoglobinopatie nepodstoupila. Na naše doporučení byla indikována genetická konzultace a v 9. týdnu těhotenství bylo provedeno vyšetření karyotypu obou partnerů. Pomocí DNA sekvenování byla identifikována heterozygotní forma mutace β-talasémie s mutací IVS I-6. T>C, dle HGVS HBB: c.92+6T–>C, fenotyp β-plus u obou probandů. Na základě doporučení genetika i při negativním prvotrimestrálním screeningu vrozených vývojových vad ve 13. dokončeném týdnu těhotenství se pacientka rozhodla podstoupit invazivní prenatální diagnostiku, odběr choriových klků. Ze získaného materiálu byla u plodu potvrzena homozygotní mutace β-talasémie, která se fenotypově může projevovat jako β-thalassemia intermedia až β-thalassemia maior. Oba partneři se po další konzultaci s genetikem na základě uvedených výsledků rozhodli pro ukončení těhotenství. V 15. dokončeném týdnu těhotenství podán misoprostol vaginálně a následně dinoprost in utero pod ultrasonografickou kontrolou. Po potratu plodu provedena revize děložní dutiny v celkové anestezii a antibiotické cloně. Pacientka po psychologickém otřesu a abortu další těhotenství zatím neplánuje a přála si perorální hormonální antikoncepci, k jejímuž nasazení nebyly nalezeny kontraindikace.
Výsledky krevního obrazu pacientky v těhotenství s nálezem hypochromní mikrocytární anémie, kompenzační erytrocytózou a normálními hodnotami železa i transferinu. Vzhledem k tomu, že byla pacientka vyšetřena v těhotenství genetikem, nebyla použita metoda vyšetření retikulocytů. Pokud by jejich počet byl normální, lze talasémie jednoduše odlišit od hemolytických anémií.
BIOCHEMIE
S_Železo: 18,9 µmol/l; S_Ferritin: 25 µg/l; S_Transferrin: 2,6 g/l; HEMATOLOGIE B_Leukocyty: 6,64 109/l; B_Erytrocyty: 5,44 1012/l; B_Hemoglobin: 116 g/l; B_Hematokrit: 0,376 1; B_MCV: 69,1 fl; B_MCH: 21,3 pg; B_MCHC: 309 mm g/l; B_RDW-CV: 16,1 %; B_Trombocyty: 222 109/l; B_Trombokrit: 0,025 10–3; B_PDW: 16,3 fl; B_MPV: 11,4 fl.
DISKUZE
Celková prevalece přenašečů β-talasémie na iberském poloostrově se předpokládá u 1,5 % populace, což odpovídá celosvětové prevalenci. Naopak v jihovýchodní Asii (kam patří i Vietnam, Laos, Thajsko atd.) je možné diagnostikovat až 60 druhů talasemických syndromů s různou prevalencí přenašečů; 9 % přenašečů beta β-talasémie, 8 % srpkovité anémie, až 60 % populace jsou přenašeči pro HbE talasémie [6].
I když Portugalsko nepatří do endemického talasemického pásu a distribuce přenašečů β-talasémie je zde nejnižší z oblasti Středozemí, naše pacientka i její partner jsou heterozygotní přenašeči s pravděpodobností β-thalassemia maior u potomků 25 %. Migrace zvýšila prevalenci β-thalassemia maior v zemích, kde se dříve tato onemocnění nevyskytovala. Efektivní screening v rizikových zemích však dokázal počet homozygotních případů účinně zredukovat (např. Řecko, Turecko, Kypr). Dle zprávy WHO v evropských státech zatím chybí jednotný screeningový nebo registrační systém heterozygotních jedinců [7].
Zásadní úlohu v diferenciální diagnostice hemoglobinopatií má hematolog. Naše pacientka ani partner nevyužili před koncepcí možnosti vyšetření na hematologii, kde by pomocí vyšetření periferní krve byla provedena diagnostická rozvaha. Hematologické vyšetření zahrnuje kompletní analýzu krevního obrazu, vč. počtu retikulocytů, krevního nátěru pro stanovení morfologie erytrocytů a kapilární elektroforézu hemoglobinu. Kapalinovou chromatografií lze detekovat pacienty s abnormálním hemoglobinovým spektrem. Genetická analýza slouží k určení typu mutace. Při detekci nosičství talasemie u ženy a jejího partnera plánujících těhotenství (nebo už těhotné ženy) je nutné v rámci prenatální diagnostiky vyloučit homozygotní nebo dvojitě heterozygotní postižení plodu.
S možností prekoncepční diagnostiky hemoglobinopatií a prenatální diagnostikou se zaměřením na heterozygotní i homozygotní formy se v české literatuře bohužel příliš nesetkáme. Ambulantní gynekologové by měli mít přehled o diferenciální diagnostice mikrocytárních anémií. Pomocí velice jednoduchého screeningového dotazníku je možné pátrat po hemoglobinopatiích u párů z endemického pásu. Lze použít např. Britský dotazník NHS o rodinném původu, a to i na klinikách asistované reprodukce [8].
U těhotné s β-thalassemia minor zvažujeme suplementaci železem jen v případě průkazu jeho nedostatku. Může totiž docházet k jeho relativnímu nadbytku a většinou nepřinese efektivní terapeutický účinek. Při hodnotě hemoglobinu pod 100 g/l lze zvážit prepartální transfuzi. Pacientky s β-talasémií mají vyšší riziko rozvoje gestačního diabetu. Pokud dojde k anémii z hypervolemie a hladina hemoglobinu je nízká již v prvním trimestru, zvyšuje se riziko intrauterinní růstové restrikce, nízké porodní váhy a předčasného porodu. Časná anémie negativně ovlivňuje vaskularizaci a angiogenezi v placentě [9].
Ze zkušeností zemí s vysokou migrací nebo z endemického pásu je prokázáno, že preimplantační genetická diagnostika u heterozygotního páru je úspěšnou metodou umožňující vyhnutí invazivnímu prenatálnímu testování (kordocentéza, biopsie choriových klků, amniocentéza a následná indukce abortu z genetické indikace a ukončení těhotenství), které může být pro pár traumatizující. Úspěšnost implantace embrya je však při jednom IVF cyklu s PDG okolo 30 % [10]. V izraelské studii je celoživotní terapie jedince s thalassemia maior odhadována na 2 miliony dolarů, oproti 63 tisícům dolarů za preventivní opatření s cílem zachytit postiženého jedince [11]. Díky preimplantační genetické diagnostice lze selektovat zdravá embrya a zvýšit šanci na porod zdravého jedince nebo heterozygota s minimálními příznaky. Páry s prokázanou β-thalassemia minor ve věku nad 30 let je vhodné poučit, že časnější provedení IVF cyklu s preimplantační diagnostikou embryí může zlepšit implantační potenciál vitrifikovaných embryí, který i o několik let později stále odpovídá věku ženy při odběru oocytů. Možností je také IVF/ICSI (intracytoplasmic sperm injection) s použitím spermatu zdravého dárce. Neinvazivní prenatální testování z periferní krve pacientky od 9. týdne těhotenství nabízí v posledních letech rozšířenou možnost detekce hemoglobinopatií. Heterozygotní páry by také měly být o této variantě poučeny [12].
V České republice jsou pacienti soustředěni do Hematologických center a Center pro vzácné choroby. Do roku 2017 byla na základě testování v ÚHKT vyslovena podezření na β-talasémii u 214 nemocných, z toho bylo 70 nemocných českého původu. Diagnóza byla ověřena molekulárně geneticky u 49 nemocných. V ÚHKT byli sledováni 2 nemocní s kombinací heterozygotních forem β-talasémie s HbE s klinickým obrazem thalassemia intermedia až major. Nemocní trpí těžkým stupněm anémie (Hb 50–86 g/l), jsou trvale závislí na podávání transfuzí erytrocytů a podávání chelatačních látek v poměrně vysokých dávkách. U obou nemocných jsou přítomny malformace skeletu a retardace růstu. U obou byla již v dětství provedena splenektomie, ale s poměrně malým efektem na redukci počtu transfuzí [13].
ZÁVĚR
Primární a sekundární prevence talasémií se díky globalizaci posouvá i do zemí, které nebyly původně endemické. Screening heterozygotů je jedinou efektivní metodou, jak snížit riziko narození jedince s β-thalassemia maior. Důkladně odebraná anamnéza je základem každého klinického vyšetření. Dále je nutná multioborová spolupráce gynekologa s hematologem a genetikem. Genetické poradenství je jednou z nejefektivnějších forem prevence závažných forem, přesná genetická diagnostika je však komplikovaná, jelikož bylo popsáno více než 300 β-talasemických mutací s variabilním fenotypem [14]. Z důvodu prenatální diagnostiky nosičství talasémie je však dostačující přesná anamnéza a vyšetření krevního obrazu, hodnot sérového železa a feritinu a elektroforéza hemoglobinu A2 a F na pracovišti se zkušeností s diagnostikou hemoglobinopatií (ÚHKT Praha a HOK FN Olomouc).
Nezřídka může situaci komplikovat kulturní, jazyková nebo náboženská bariéra, etické aspekty a obtížná komunikace. U párů, které si nepřejí těhotenství a oba rodiče jsou nosiči talasemické alely, pečlivě plánujeme koncepci nebo volíme spolehlivou kontracepci v době, kdy těhotenství není aktuální.
Homozygotní hemoglobinopatie závažně ovlivňují kvalitu života postiženého jedince, mají silný socio-ekonomický dopad na celou rodinu, dítě i na zdravotní systém. Novou nadějí pro homozygotní jedince závislé na transfuzní terapii je terapie pomocí genetické manipulace buněk ex vivo s následnou transplantací [14]. Zdravotní systém by se měl rychle adaptovat na celosvětové změny, zvyšovat informovanost profesionálů, zabývat se účinnou prevencí, registrací nosičů a zvážit cílený screening přenašečů hemoglobinopatií u rizikové populace.
PODÍL AUTORŮ NA PŘÍPRAVĚ RUKOPISU
JR – příprava rukopisu
MM – korekce rukopisu
ČESTNÉ PROHLÁŠENÍ
Autoři práce prohlašují, že v souvislosti s tématem, vznikem a publikací tohoto článku nejsou ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou.
Doručeno do redakce dne: 3. 6. 2022.
Přijato po recenzi dne: 15. 8. 2022.
MUDr. Jana Racková
Gynekologicko-porodnická klinika
1. LF UK a Nemocnice Na Bulovce,
Budínova 2
182 00 Praha 8
e-mail: endowomen@gmail.com
Literatura
1. Indrák K, Divoká M, Pospíšilová D, et al. Hemoglobinopatie. Vnitr Lek. 2018; 64(5): 476–487.
2. Galanello R, Origa R. Beta-thalassemia. Orph J Rare Dis. 2010; 5: 11.
3. Weatherall DJ. Phenotype-genotype relationships in monogenic disease: lessons from the thalassaemias. Nat Rev Genet. 2001; 2(4): 245–255.
4. Shang X, Xu X. Update in the genetics of thalassemia: What clinicians need to know. Best practice & research. Clin Obst Gynaecol. 2017; 39: 3–15.
5. Galanello R, Cao A. (1998). Relationship between genotype and phenotype. Thalassemia intermedia. Ann New York Acad Sci. 1998; 850: 325–333.
6. Fucharoen S, Winichagoon P. Haemoglobinopathies in Southeast Asia. Indian J Med Res. 2011; 134(4): 498–506.
7. Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull WHO. 2008; 86(6): 480–487.
8. Publikováno elektronicky: Family origin questionnaire. https: //assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/830289/SCT_Family_Origin_Questionnaire.pdf.
9. Kadyrov M, Kosanke G, Kingdom J, Kaufmann P. Increased fetoplacental angiogenesis during first trimester in anaemic women. Lancet. 1998; 352(9142): 1747–1749.
10. Monni G, Peddes C, Iuculano A, Ibba RM. From prenatal to preimplantation genetic diagnosis of β-thalassemia. Prevention model in 8748 cases: 40 years of single center experience. J Clin Med. 2018; 7(2): 35.
11. Traeger-Synodinos J. Preimplantation genetic diagnosis, an alternative to conventional prenatal diagnosis of the hemoglobinopathies. Int J Lab Hematol. 2013; 35(6): 571–579.
12. Chen C, Li R, Sun J, et al. Noninvasive prenatal testing of α-thalassemia and β-thalassemia through population-based parental haplotyping. Genome Med. 2021; 13(1): 18.
13. Čermák J, Válka J, Vostrý M, Škranc S, Beličková M. Centrum pro vzácné choroby červené krevní řady v Ústavu hematologie a krevní transfuze. Transfuze Hematol Dnes. 2017; 23(Suppl 1): 76–86.
14. Aprile A, Sighinolfi S, Raggi L, Ferrari G. Targeting the hematopoietic stem cell niche in β-thalassemia and sickle cell disease. Pharmaceuticals. 2022; 15(5): 592.
Článek byl uveřejněn s laskavým svolením redakce Transfuze a hematologie dnes.
www.prolekare.cz/casopisy/transfuze-hematologie-dnes