Muscle pain as a rare autoimmune symptom of monoclonal IgM gammopathy – case report and overview of autoimmune manifestations associated with monoclonal immunoglobulins
Král Z., Krejčí M., Pour L., Adam Z.
Interní hematologická a onkologická klinika LF MU a FN Brno
Transfuze Hematol Dnes 2021; 27(1): 57–61. doi: 10.48095/cctahd202157
SOUHRN: Následující sdělení popisuje vzácný případ Waldenströmovy makroglobulinémie s autoimunitní myopatií a dále stručný přehled možných dalších autoimunitních projevů přítomnosti monoklonálních imunoglobulinů (Mig). Waldenströmova makroglobulinémie se u pacientky manifestovala bolestmi svalů a anémií. Potíže trvaly asi 3 roky před stanovením diagnózy WM v roce 2014 se zhoršením stavu v posledním roce, kdy bolesti postihovaly nejen zádové svaly, ale i svaly končetin. Hodnoty kreatinkinázy (CK) a myoglobinu byly výrazně zvýšené. Podezření na polymyositidu nebylo potvrzeno. Byla prokázána monoklonální gamapatie typu IgM a lymfoplazmocytární lymfom v kostní dřeni. Proto byla zahájena léčba ve složení rituximab, cyklofosfamid, dexametazon. Celkem bylo podáno 8 cyklů v období 7/2014–2/2015. Již po pátém cyklu bylo dosaženo negativní imunofixace. normalizovaly se hodnoty CK, myoglobinu a hemoglobinu a také došlo k ústupu myalgií. V prosinci 2019 stále trvá kompletní remise WM s normálními či mírně zvýšenými hodnotami CK a myoglobinu.
SUMMARY: This article describes a rare case of Waldenström’s macroglobulinemia with autoimmune myopathy and presents a short overview of other possible autoimmune manifestations associated with the presence of monoclonal immunoglobulin (MIg). In our patient, Waldenström’s macroglobulinemia manifested as chronic muscle pain and anaemia. The patient suffered from back muscle pains for about 3 years before being diagnosed with WM, with progression of myalgia and pain of the upper and lower limbs in the last year prior to diagnosis. Serum myoglobin and creatinkinase (CK) levels were significantly increased. Polymyositis was not confirmed. The diagnosis of WM was established in 2014, with typical infiltration with lympho-plasmacytoid lymphoma in the bone marrow and the presence of IgM lambda paraproteinemia in the serum. The patient started treatment with the anti-CD20 sssmonoclonal antibody rituximab, cyclophosphamide and dexamethasone and received 8 cycles from 7/2014–2/2015. The IgM disappeared, the immunofixation results normalised as did myoglobin, CK and haemoglobin after the 5th cycle and there was significant improvement of muscle pain. The patient achieved a complete remission with no signs of relapse/progression at the last follow-up in 12/2019 with normal or only mildly elevated CK and myoglobin.
ÚVOD
Waldenströmova makroglobulinémie (WM) je vzácné B-lymfoproliferativní onemocnění s incidencí 3–4 případy/1 milion obyvatel, medián věku v době diagnózy je 63–75 let [1]. Diagnóza WM vyžaduje průkaz lymfoplazmocytárního lymfomu (LPL) v kostní dřeni a dále přítomnost monoklonálního imunoglobulinu (MIg) typu IgM v séru. U WM je vždy přítomno postižení kostní dřeně, méně často i lymfatických uzlin a sleziny [1]. Klinické příznaky u pacientů s WM mohou být velmi různorodé, jsou způsobeny infiltrací kostní dřeně s potlačením fyziologické krvetvorby, dále možnou extramedulární proliferací a také přítomností MIg, která může způsobit mimo jiné periferní neuropatii, hyperviskozitu, kryoglobulinémii nebo nemoc chladových aglutininů. Dále u WM mohou být přítomny některé paraneoplastické projevy (např. B-symtomy, kožní a krvácivé projevy) [1,2].
Monoklonální imunoglobulin IgM obvykle charakterizuje masu klonálních plazmocytů či lymfoplazmocytů, takže změny jeho koncentrace slouží jak ke sledování této nemoci, tak k hodnocení léčebné odpovědi u WM. U malé části osob s přítomností Mig typu IgM se můžeme setkat s vazbou této protilátky na některé organizmu vlastní antigeny, a tedy s projevy autoimunity. Monoklonální imunoglobuliny typu IgM způsobují autoimunitní projevy častěji než Mig typu IgG nebo IgA. Snad nejvíce známé komplikace, které může způsobit přítomnost MIg, jsou koagulopatie a neuropatie [1,3]. Oproti tomu je myopatie při IgM gamapatii popsána v odborné literatuře jen velmi ojediněle, většinou formou popisů jednotlivých případů [4–10]. V následujícím sdělení se věnujeme popisu vzácného souběhu WM a autoimunitní myopatie a dále stručnému přehledu možných dalších autoimunitních projevů monoklonálních imunoglobulinů.
VLASTNÍ POPIS PŘÍPADU
Pacientka narozená v roce 1955 měla ve 23 letech zjištěné bronchiální astma a hypofunkci štítné žlázy, pravděpodobně autoimunitního původu. S ničím dalším se pravidelně neléčila. Nespecifické myalgie stěhovavého charakteru především v oblasti svalstva zad si uvědomovala již od roku 2010. Bolesti svalů se v roce 2013 postupně rozšířily i na končetiny, dále se objevila slabost a mírná únava, především v závislosti na fyzické zátěži. Pacientka byla pro únavové stavy a bolesti svalů zprvu odeslána na spádovou revmatologickou ambulanci, zde byly v rámci celkového vyšetření zjištěny patologické laboratorní hodnoty, především zvýšení koncentrace myoglobinu v séru, zvýšení aktivity kreatinkinázy (CK) a laktátdehydrogenázy (LD), dále byla přítomna mírná mikrocytární anémie s hemoglobinem (Hb) 110 g/l, zvýšená hodnota sedimentace a přítomnost Mig typu IgM lambda v séru v nízké koncentraci kolem 12 g/l. Bylo vysloveno podezření na suspektní polymyozitidu na podkladě svalových bolestí, mírné slabosti a nálezu zvýšených hodnot svalových enzymů a nemocná byla v roce 2013 odeslána k přešetření za hospitalizace do Revmatologického ústavu v Praze. Pacientka udávala zvýšenou únavu, dále bolesti svalů stěhovavého charakteru, minimální svalovou slabost, nebyly přítomny obtíže s polykáním nebo dýcháním. Objektivně byla přiměřená svalová hmota bez hypotrofie, pacientka byla bez známek artritidy, nebyl přítomen kožní exantém. Při zobrazovacích vyšetřeních nebyla zjištěna lymfadenopatie či organomegalie. Laboratorně byly přítomny zvýšené hodnoty myoglobinu, CK a LD při kompletně negativním imunologickém vyšetření dle doporučení revmatologa (CIK, C3, C4, autoprotilátky typu ANA, ENA, ds-DNA, antikardiolipinové protilátky, anti-CCP, revmatoidní faktor a další). U pacientky byl dále proveden svalový test a také elektromyografické vyšetření (EMG) bez patologického nálezu. Dále byla provedena i svalová biopsie se závěrem minimálních strukturálních změn charakteru „minimal change myopathy“. Exprese spektrinu, merosinu, emerinu, sarkoglykanů dystroglykanů byla imunohistochemicky v normě, histopatologem byla provedena řada doplňujících vyšetření (imunohistochemických, imunofluorescenčních a molekulárně-genetických) bez průkazu zásadní patologie, nebyla morfologicky prokázána metabolická či zánětlivá myopatie. Stav byl uzavřen jako ne zcela jasný, s tím, že diagnóza polymyozitidy při absenci svalové slabosti, s normálním výsledkem EMG a negativní imunologií není pravděpodobná. Vzhledem ke zjištěné monoklonální gamapatii typu IgM bylo revmatology doporučeno další vyšetření na odborném hematologickém pracovišti.
Při prvním vyšetření na naší hematologicko-onkologické ambulanci v roce 2013 udávala pacientka difúzní bolesti ve svalstvu zad, dále bolesti svalů horních i dolních končetin a zvýšenou únavu. Pacientka neudávala teploty ani noční pocení, měla stabilní hmotnost, krvácivé projevy nebyly přítomny. Při vstupním vyšetření v 11/2013 byl objektivní nález bez průkazu výraznější patologie, při laboratorním vyšetření byla zjištěna anémie s koncentrací Hb 107 g/l, ostatní parametry krevního obrazu byly v normě (počty trombocytů a leukocytů). V séru byl Mig typu IgM lambda v koncentraci 14,7 g/l. Byly zvýšené hodnoty myoglobinu a CK, vstupní hodnota myoglobinu byla 250 µg/l (norma 25–58 µg/l) a hodnota CK byla 12 ukat/l (norma 0,43–2,34). Základní parametry biochemického vyšetření byly v normě (urea, kreatitin, bilirubin a jaterní enzymy, ionty, C-reaktivní protein, celková bílkovina a albumin). U pacientky bylo provedeno vyšetření kostní dřeně (trepanobiopsie), histologicky byla v kostní dřeni detekována až 30 % lymfoplazmocytární infiltrace v nodulárních, non-paratrabekulárních agregátech. Imunohistochemicky se jednalo o CD 20+, CD5–, CD23–, cyklinD1–, IgM lambda+ lymfoplazmocytoidní buňky. V pozadí byly reaktivní mastocyty. Patolog tento nález hodnotil jako obraz infiltrace kostní dřeně lymfoplazmocytárním lymfomem. U pacientky bylo vstupně provedeno PET/CT vyš. bez průkazu zásadní patologie. Symptomy nemoci byly v tomto případě především progredující myalgie stěhovavého charakteru a zvýšená únava. Vyjma patologické infiltrace kostní dřeně při lymfoplazmocytárním lymfomu nebyla odhalena jiná příčina obtíží.
U pacientky byla zahájena kombinovaná terapie ve složení rituximab (monoklonální protilátka anti-CD20), cyklofosfamid a dexametazon (režim DRC). Rituximab byl podáván v dávce 375 mg/m2 1× měsíčně. Dexametazon byl podáván v dávce 20 mg/den 1.–4. den a 15.–18. den ve 28denním cyklu. Cyklofosfamid byl podáván v dávce 500 mg/m2 jako intravenózní infuze 1. a 15. den 28denního cyklu. Celkem bylo podáno 8 cyklů této terapie v období 8/2014–3/2015.
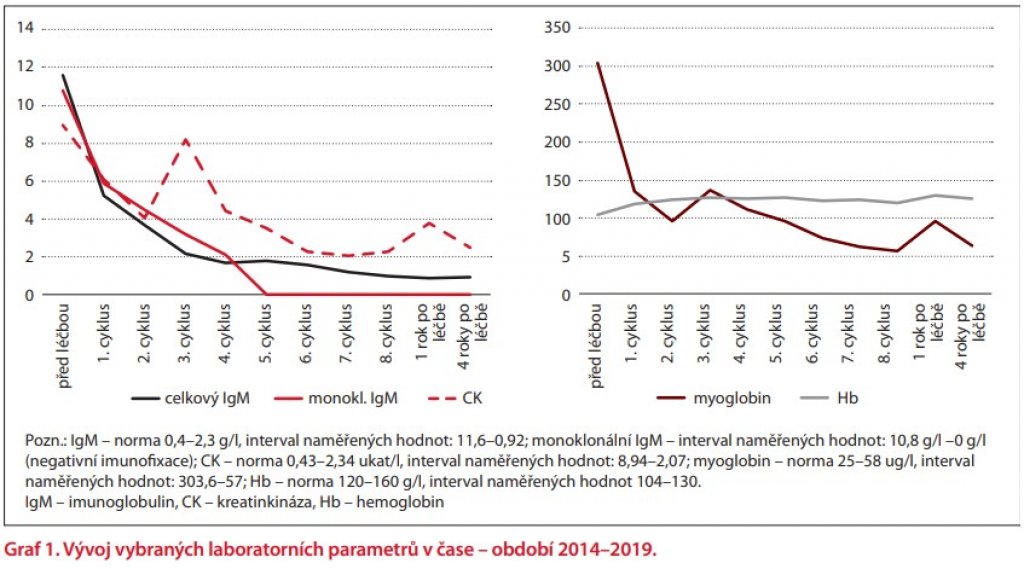
V průběhu léčby byl zřetelný signifikantní pokles hodnot myoglobinu a hodnot CK. Vývoj vybraných laboratorních parametrů v období 2014–2019 (před léčbou, v průběhu 8 cyklů DRC a po léčbě) přehledně zobrazuje graf 1.
Již po aplikaci 5 cyklů DRC bylo dosaženo kompletní remise nemoci s negativní imunofixací, subjektivní příznaky nemoci se minimalizovaly, bolesti svalů prakticky ustoupily, došlo k normalizaci hodnoty Hb, dále k významnému poklesu hodnot myoglobinu a CK do oblasti normálních či pouze lehce nadlimitních hodnot. Pacientka absolvovala celkem 8 cyklů DRC a nadále je ambulantně dlouhodobě sledována v několikaměsíčních intervalech prozatím bez nutnosti další terapie. Průběh terapie DRC byl u pacientky bez komplikací a nebyly zaznamenány žádné výraznější nežádoucí účinky léčby, pacientka tuto terapii zvládla plně ambulantně. Při kontrole v prosinci 2019 byla pacientka nadále ve stabilizovaném klinickém stavu, subjektivně bez výraznějších obtíží, bez projevů myopatie, při laboratorním vyšetření zůstává imunofixace MIg IgM negativní, jsou přítomné pouze lehce nadlimitní hodnoty CK (2,49 µkal/l) a myoglobinu (63,6 µg/l) a krevní obraz je normální.
DISKUZE
Monoklonální imunoglobuliny jsou obvykle produkovány jedním klonem plazmocytů (Mig nejčastěji typu IgG, nebo IgA) anebo lymfoplazmocytárních buněk (MIg typu IgM). Ve většině případů se tyto MIg nevážou na žádné tělu vlastní antigeny, u malé části nemocných však k této vazbě může docházet a následně mohou způsobovat klinické projevy autoimunity.
Je zjištěno, že se na autoantigeny vážou MIg typu IgM podstatně častěji než ostatní typy. Proto jsou také monoklonální gamapatie nejistého významu typu IgM a WM provázeny autoimunitními projevy častěji než u jiných typů monoklonálních gamapatií [11,12].
Průkaz vazby MIg na autoantigeny však nemusí vždy znamenat klinicky zřetelné symptomy. Četnost klinicky zřetelných autoimunitních chorob způsobených MIg je nižší než četnost průkazu přítomnosti MIg vázajícího se na autoantigeny. To bylo prokázáno cílenou analýzou přítomnosti monoklonálních IgM autoprotilátek [13,14].
Jak častá je vazba monoklonální IgM protilátky na autoantigeny?
Carlizzi et al. uvádí, že vazbu MIg typu IgM na nukleární antigeny detekovali u jedné třetiny vyšetřených osob [14]. Pokud byla analyzována podskupina osob starších 60 let, tak průkaz monoklonální protilátky typu IgM s vazbou na nukleární antigeny byl u 40 % případů [14]. Klinické symptomy odpovídající přítomnosti antinukleárních protilátek byly detekovány pouze u jedné osoby, pozitivita průkazu antinukleárních protilátek byla závislá na volbě testu pro jejich průkaz [14].
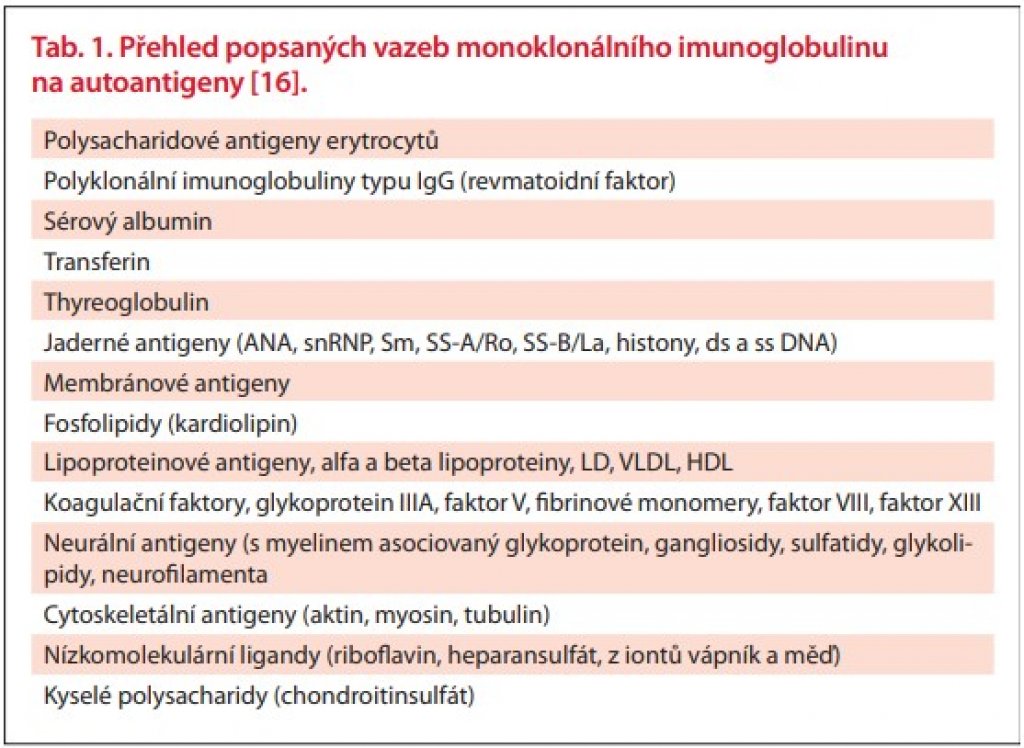
Vazba MIg typu IgM na jeden či více autoantigenů byla prokázána u 71 % ze 172 osob s IgM monoklonální gamapatií. Jednalo se o analýzu velmi širokého spektra autoprotilátkové aktivity. Vazbu MIg na jaderné antigeny (antinukleární protilátky) mělo 11 % vyšetřených [15]. V této práci ale není uvedeno, jak časté byly klinické projevy těchto autoprotilátek. Uvedené práce tedy signalizují, že u pacientů s MIg, zvláště jedná-li se o imunoglobulin typu IgM, musíme vždy myslet na možnost jeho autoimunitní aktivity. Stone et al. uvádí přehled antigenů, na které byla prokázána vazba MIg [16]. Jsou to sumarizované informace z různých publikací, které popisovaly vazbu MIg u osob s WM a s mnohočetným myelomem. V tab. 1. chceme ilustrovat, že vyjma relativně známých projevů vazby MIg na autoantigeny, jako jsou např. nemoc chladových aglutininů, smíšená kryoglobulinémie a neuropatie, se můžeme setkat s klinickými důsledky vazby na kterýkoli z antigenů uvedených v tabulce vč. antigenů svalových vláken.
Jak častá je klinická manifestace autoimunitní aktivity monoklonálního imunoglobulinu?
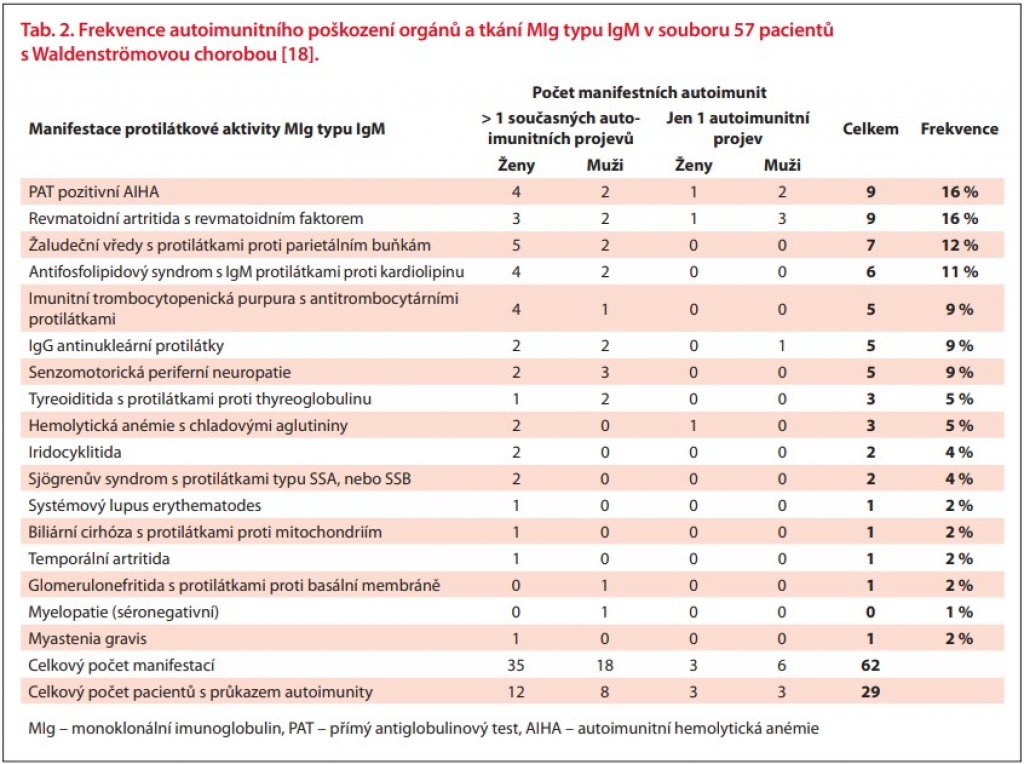
V literatuře lze nalézt dvě práce hodnotící výskyt autoimunitních poruch u pacientů s WM. V malém souboru 12 pacientů s WM byly autoimunitní projevy prokázány u více než u poloviny pacientů (57 %). Jednalo se o Hashimotovu tyreoiditidu, perniciózní anémii, imunitní trombocytopenii, nemoc chladových aglutininů, chronickou demyelinizační neuropatii, temporální arteritidu a další [17]. Podobné údaje, ale ve větším souboru celkem 57 pacientů s WM, byly publikovány již v roce 1999 [18]. Výsledky této práce uvádíme v tab. 2. V souboru 57 pacientů s WM byly nejčastější Coombs pozitivní autoimunitní hemolytická anémie (AIHA) (16 %), dále séropozitivní revmatoidní artritida (16 %) a žaludeční vřed spojený s přítomností protilátky proti parietálním buňkám bez přítomnosti perniciózní anémie (12 %) [18]. Manifestace autoimunity byla prokázána u 29 z 57 vyšetřovaných, z toho ve 20 případech se současně vyskytly nejméně dva autoimunitní projevy a v 9 případech jeden autoimunitní projev. Jedna manifestace byla častější u mužů, více než jedna byla častější u žen [18].
Myopatie při monoklonální gamapatii
S postižením svalové tkáně při monoklonální gamapatii jsme se na našem pracovišti setkali v průběhu 20 let práce na naší hematoonkologické ambulanci poprvé. Při pohledu do literatury je zřejmé, že tento typ poškození organizmu MIg je popisován i ve světové literatuře relativně vzácně, jedná se většinou o popisy jednotlivých případů. Jak dokládá náš případ i citovaná literatura, může MIg ve velmi vzácných případech poškozovat příčně pruhovanou svalovinu [4–10]. Vzácně byly popsány i autoimunitní projevy způsobené imunoglobulinem typu IgG při WM [19].
Vazbu MIg na svalová vlákna nemáme laboratorně prokázanou. Nicméně z faktu, že při poklesu koncentrace MIg při léčbě došlo jak ke zvýšení koncentrace Hb a vymizení anémie, tak k poklesu koncentrace CK a myoglobinu, usuzujeme na možnou kauzální souvislost mezi přítomností MIg a myopatií.
Léčba Waldenströmovy makroglobulínémie
Léčba WM se postupně vyvíjí na základě výsledků řady klinických studií a odborných doporučení národních i mezinárodních, která jsou opakovaně aktualizována [2,20–22]. V současné době máme k dispozici recentní odborné doporučení z roku 2019 pro diagnostiku i terapii WM [1].
Režim DRC je dobrou léčebnou alternativou pro nově léčené pacienty s malou nádorovou masou, nevyžadující rychlý nástup léčebné odpovědi. V našem případě jsme použili mírně modifikovaný režim DRC s velmi dobrým efektem a dobrou léčebnou tolerancí. Vyšší dávka dexametazonu a vyšší počet léčebných cyklů (z 6 na 8) vedly k dosažení velmi dobré dlouhodobě přetrvávající léčebné odpovědi a prakticky k vymizení klinických obtíží pacientky.
ZÁVĚRY PRO PRAXI
Monoklonální imunoglobulin typu IgM může vyvolávat velmi pestré autoimunitní projevy. Poškození příčně pruhovaných svalů je jednou z velmi vzácných projevů přítomnosti monoklonálního imunoglobulinu.
Diferenciální diagnostika nejasného poškození orgánů a tkání je velmi široká. V případě průkazu přítomnosti monoklonálního imunoglobulinu typu IgM (imunofixační elektroforézou) by mělo být pomýšleno i na diagnózu WM a možnou vzácnou souvislost s autoimunitními projevy. Při adekvátní terapii může dojít k významné stabilizaci klinického stavu pacienta a k vymizení autoimunitních, ale i dalších klinických projevů.
Podíl autorů na přípravě rukopisu
KZ – příprava rukopisu
KM, PL, ZA – korekce a revize rukopisu
Čestné prohlášení autorů
Autoři práce prohlašují, že v souvislosti s tématem, vznikem a publikací tohoto článku nejsou ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou.
Do redakce doručeno dne 14. 2. 2020.
Přijato po recenzi dne 3. 7. 2020.
prof. MUDr. Zdeněk Adam, CSc.
Interní hematologická a onkologická klinika
LF MU a FN Brno
Jihlavská 20, 625 00 Brno
e-mail: adam.zdenek@fnbrno.cz
Literatura
1. Kaščák M, Hájek R, Minařík J, et al. Diagnostika a léčba Waldenströmovy makroglobulinemie. Transfuze Hematol Dnes. 2019; 25(Suppl 1): 7–33.
2. Treon SP. How I treat Waldenström macroglobulinema. Blood. 2015; 126(6): 721–732.
3. Bulikova A, Smejkal P, Zavřelová J, et al. Získané inhibitory krevního srážení. Int Med Prax. 2008; 10: 336–339.
4. Al-Lozi MT, Pestronk A, Yee WC, et al. Myopathy and paraproteinemia with serum IgM binding to a high-molecular-weight muscle fiber surface protein. Ann Neurol. 1995; 37: 41–46.
5. Bakri K, Haydar AA, Davis J, et al. Waldenström‘s macroglobulinaemia presenting as isolated epistaxis: a common complaint but a rare cause. Int J Clin Pract. 2004; 58: 81–82.
6. Deconinck N, Laterre EC, Van den Bergh PY. Adult-onset myopathy and monoclonal gammopathy: a case report. Acta Neurol Belg. 2000; 100: 34–40.
7. Eymard B, Brouet JC, Collin H, et al. Late-onset rod myopathy associated with monoclonal gammopathy. Neuromuscul Disord. 1993; 3(5–6): 557–560.
8. Keller CE, Hays AP, Rowland LP, et al. Adult-onset myopathy and monoclonal gammopathy. Arch Neurol. 2006; 63(1): 132–134.
9. Bockorny B, Atienza J, Dasanu CA. Autoimmune manifestation in patients with Waldenström macroglobulinemia. Lymphoma Myeloma Leuk. 2014; 14: 456–549.
10. Stone MJ. Pathogenesis and morbidity of autoantibody syndromes in Waldenstrom‘s macroglobulinemia. Clin Lymphoma Myeloma Leuk. 2011; 11: 157–159.
11. Sakalová A, Škultétyová D, Konečná Z, et al. Súčasná klasifikácia, diagnostika a prognóza primárnych monoklonových gamapatií (paraproteinémií). Transfuze Hematol Dnes. 2010; 16: 193–201.
12. Kyle RA. Morbus Waldenström: an overview of diagnosis and therapy. Transfuze Hematol Dnes. 2008; 14(Suppl 1): 105–107.
13. Al-Lozi M, Pestronk A. Organ-specific autoantibodies with muscle weakness. Curr Opin Rheumatol. 1999; 11: 483–488.
14. Carlizzi G, Ciarla MV, Diluzio A, et al. Autoantibodies in patients with monoclonal gammopathies. Ann N.Y. Acad Sci. 2007; 1107: 206–2011.
15. Stone MJ, McElroy A, Pestronk P, et al. Human monoclonal macroglobulins with antibody activity. Semin Oncol. 2003; 30: 318–324.
16. Stone MJ, Marlini V, Pascual J. Autoantibody aktivity in Waldenström’s macroglobulinaemia. Clin Lymphoma. 2005; 5: 225–229.
17. Ansell SM. Waldenström macroglobulinemia and autoimunity: what’s the connection? Leuk Lymphoma. 2015; 6: 845–846.
18. Jonsson V, Kierkegaard A, Salling S, et al. Autoimunity in Waldenström’s macroglobulinemia. Leuk Lymphoma. 1999; 34: 373–379.
19. Nakazaki K, Hangaishi A, Nakamura F, et al. IgG-associated immune thrombocytopenia in Waldenström macroglobulinemia. Int J Hematol. 2010; 92: 360–363.
20. Maisnar V, Hájek R. Rizikové monoklonální gamapatie nejasného významu – léčit nebo neléčit? Transfuze Hematol Dnes. 2013; 19: 22–26.
21. Kaščák M, Ďuraš J, Navrátil M, et al. Autologní transplantace kmenových buněk u Waldenströmovy makroglobulinemie. Transfuze Hematol Dnes. 2016; 22: 28–38.
22. Adam Z, Hájek R, Krejčí M, et al. Diagnostika a léčba Waldenströmovy makroglobulinemie. Transfuze Hematol Dnes. 2014; 20(Suppl 1): 7–22.
Článek byl uveřejněn s laskavým svolením redakce Transfuze a hematologie dnes.
www.prolekare.cz/casopisy/transfuze-hematologie-dnes